Department of Animal Science, Faculty of Agriculture, University of Tabriz, Tabriz, Iran.
Department of Genomics, Branch for Northwest & West Region, Agricultural Biotechnology Research Institute of Iran (ABRII), Agricultural Research, Education and Extension Organization (AREEO), Tabriz, Iran.
Sci Rep. 2021 Jan 27;11(1):2367. doi: 10.1038/s41598-021-81888-z.
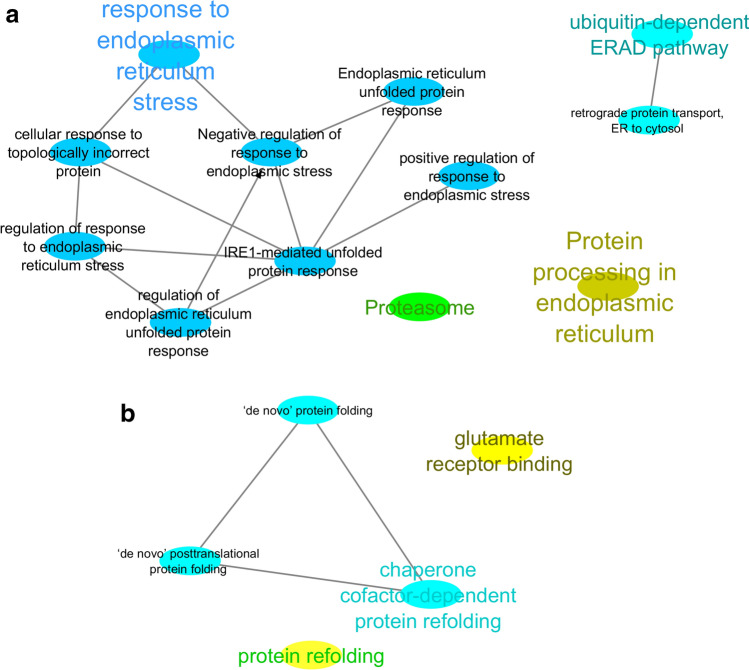
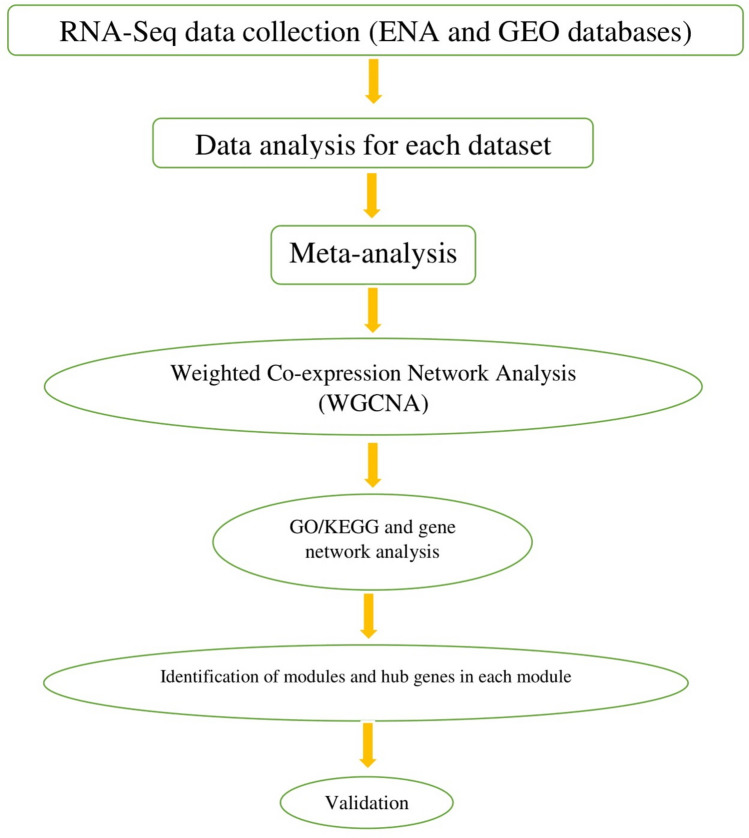
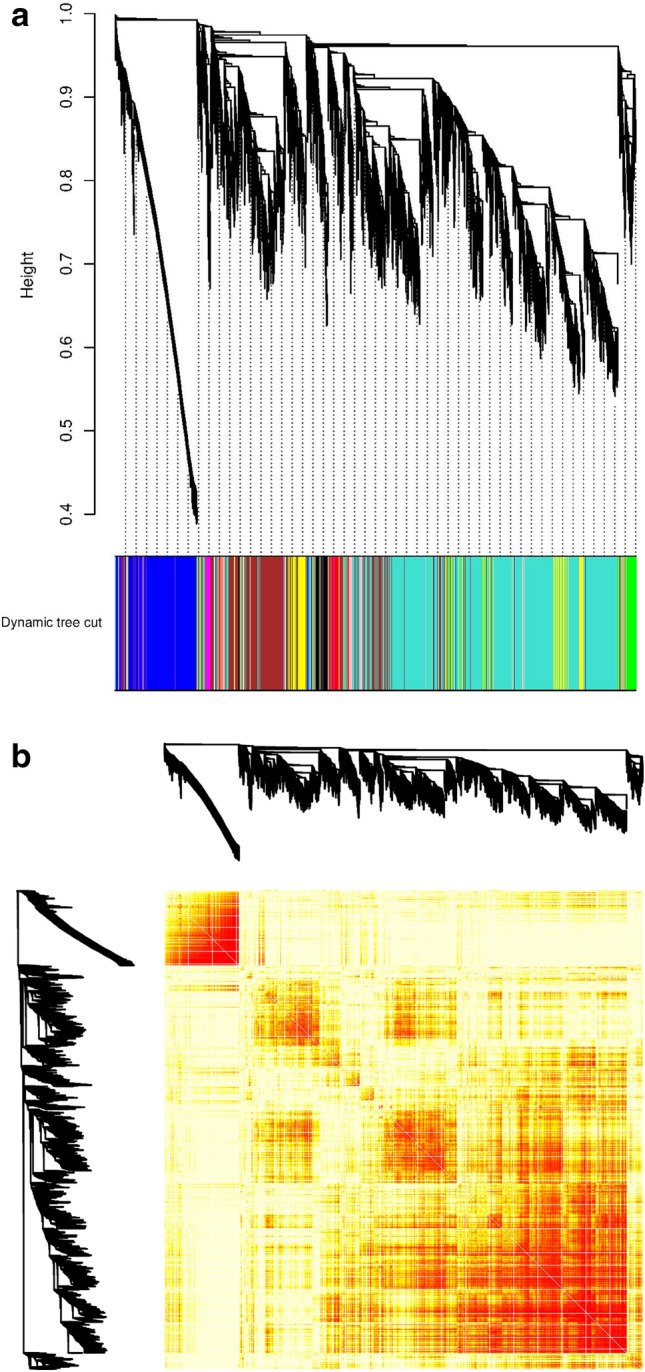
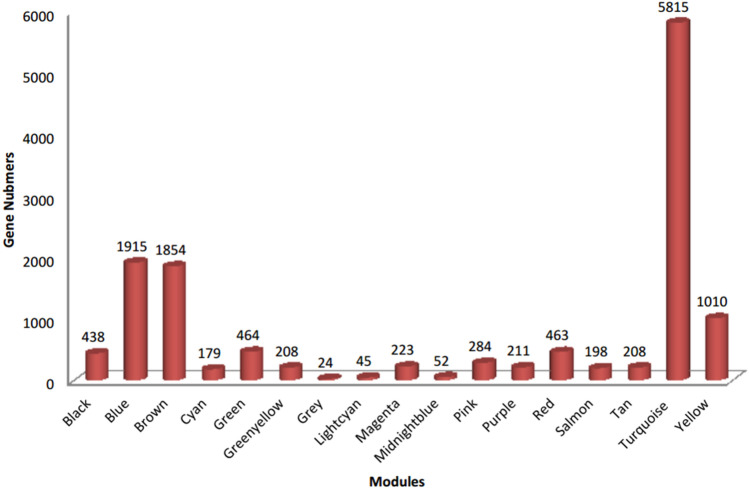
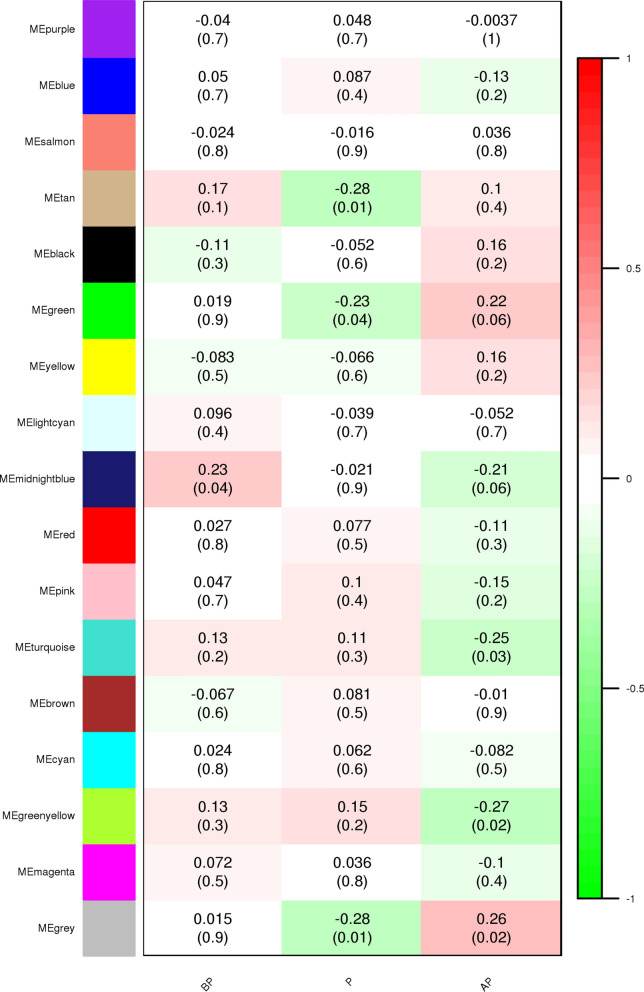
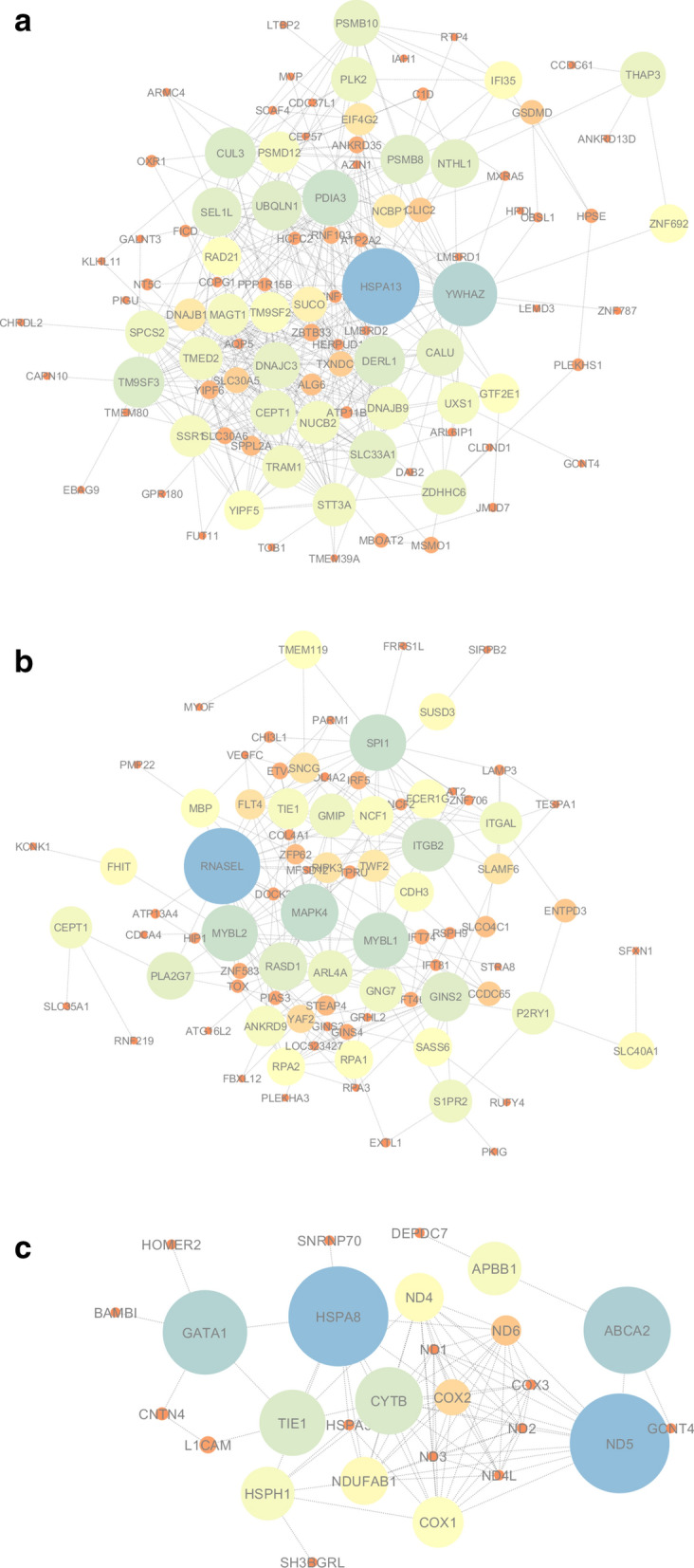
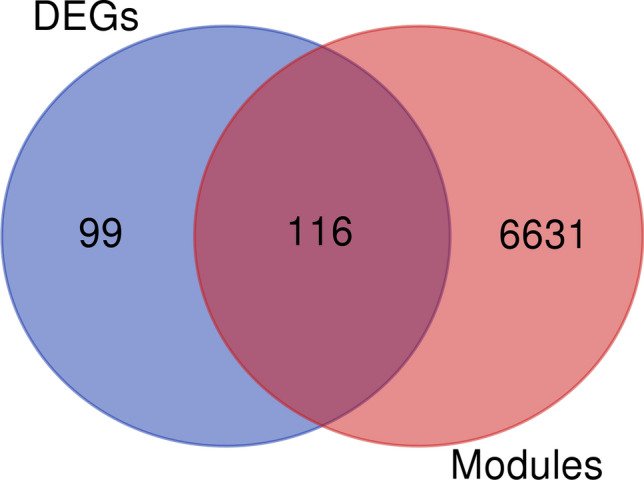
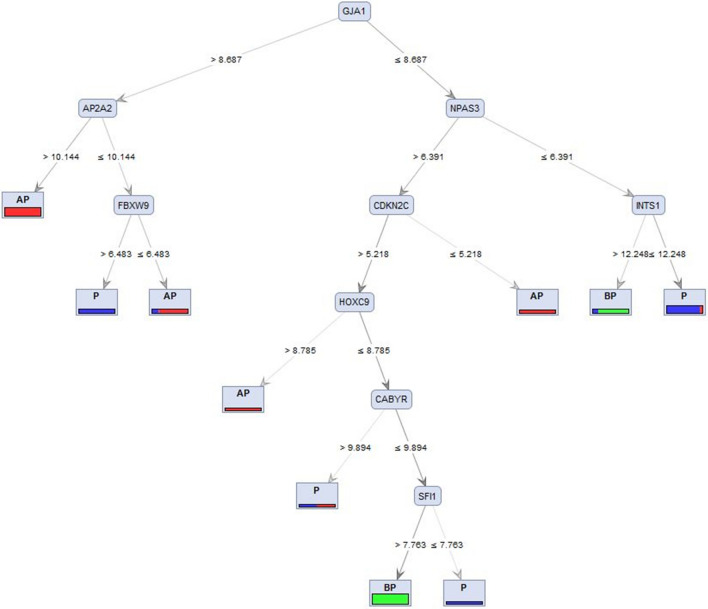
The exponential growth in knowledge has resulted in a better understanding of the lactation process in a wide variety of animals. However, the underlying genetic mechanisms are not yet clearly known. In order to identify the mechanisms involved in the lactation process, various mehods, including meta-analysis, weighted gene co-express network analysis (WGCNA), hub genes identification, gene ontology (GO), and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment at before peak (BP), peak (P), and after peak (AP) stages of the lactation processes have been employed. A total of 104, 85, and 26 differentially expressed genes were identified based on PB vs. P, BP vs. AP, and P vs. AP comparisons, respectively. GO and KEGG pathway enrichment analysis revealed that DEGs were significantly enriched in the "ubiquitin-dependent ERAD" and the "chaperone cofactor-dependent protein refolding" in BP vs. P and P vs. P, respectively. WGCNA identified five significant functional modules related to the lactation process. Moreover, GJA1, AP2A2, and NPAS3 were defined as hub genes in the identified modules, highlighting the importance of their regulatory impacts on the lactation process. The findings of this study provide new insights into the complex regulatory networks of the lactation process at three distinct stages, while suggesting several candidate genes that may be useful for future animal breeding programs. Furthermore, this study supports the notion that in combination with a meta-analysis, the WGCNA represents an opportunity to achieve a higher resolution analysis that can better predict the most important functional genes that might provide a more robust bio-signature for phenotypic traits, thus providing more suitable biomarker candidates for future studies.
知识的指数级增长导致人们对各种动物的泌乳过程有了更好的理解。然而,潜在的遗传机制尚不清楚。为了确定泌乳过程中涉及的机制,采用了各种方法,包括荟萃分析、加权基因共表达网络分析(WGCNA)、枢纽基因鉴定、基因本体论(GO)和京都基因与基因组百科全书(KEGG)途径富集分析,分别在泌乳过程的前峰(BP)、峰(P)和后峰(AP)阶段进行。基于 PB 与 P、BP 与 AP 和 P 与 AP 的比较,分别鉴定出 104、85 和 26 个差异表达基因。GO 和 KEGG 途径富集分析表明,DEGs 在 BP 与 P 和 P 与 P 中分别显著富集于“泛素依赖性 ERAD”和“伴侣辅助因子依赖性蛋白质重折叠”。WGCNA 鉴定了与泌乳过程相关的五个显著功能模块。此外,GJA1、AP2A2 和 NPAS3 被定义为所鉴定模块中的枢纽基因,突出了它们对泌乳过程的调控影响的重要性。本研究为三个不同阶段的泌乳过程复杂调控网络提供了新的见解,同时提出了几个候选基因,这些基因可能对未来的动物育种计划有用。此外,本研究支持了这样一种观点,即结合荟萃分析,WGCNA 代表了一种实现更高分辨率分析的机会,可以更好地预测可能为表型特征提供更稳健生物特征的最重要功能基因,从而为未来的研究提供更合适的生物标志物候选物。