Key Laboratory of Molecular Biophysics of the Ministry of Education, Hubei Key Laboratory of Bioinformatics and Molecular-imaging, Department of Bioinformatics and Systems Biology, Center for AI Biology, College of Life Science and Technology, Huazhong University of Science and Technology, Wuhan, 430074, Hubei, China.
BMC Genomics. 2021 Jan 28;22(1):83. doi: 10.1186/s12864-021-07372-0.
Most studies investigating human gut microbiome dynamics are conducted on humans living in an urban setting. However, few studies have researched the gut microbiome of the populations living traditional lifestyles. These understudied populations are arguably better subjects in answering human-gut microbiome evolution because of their lower exposure to antibiotics and higher dependence on natural resources. Hadza hunter-gatherers in Tanzania have exhibited high biodiversity and seasonal patterns in their gut microbiome composition at the family level, where some taxa disappear in one season and reappear later. Such seasonal changes have been profiled, but the nucleotide changes remain unexplored at the genome level. Thus, it is still elusive how microbial communities change with seasonal changes at the genome level.
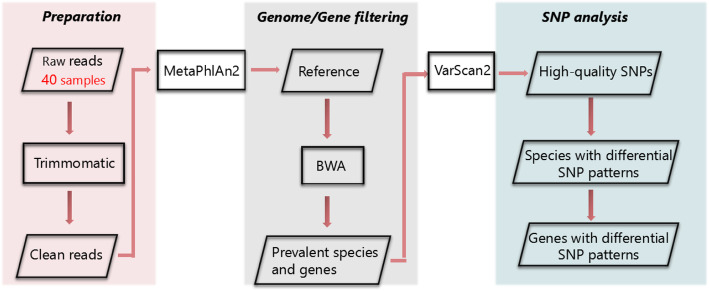
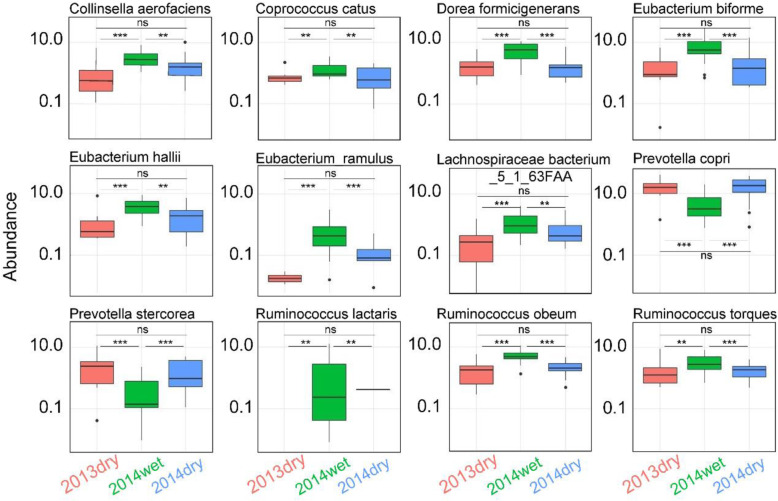
In this study, we performed a strain-level single nucleotide polymorphism (SNP) analysis on 40 Hadza fecal metagenome samples spanning three seasons. With more SNP presented in the wet season, eight prevalent species have significant SNP enrichment with the increasing number of SNP calling by VarScan2, among which only three species have relatively high abundances. Eighty-three genes have the most SNP distributions between the wet season and dry season. Many of these genes are derived from Ruminococcus obeum, and mainly participated in metabolic pathways including carbon metabolism, pyruvate metabolism, and glycolysis.
Eight prevalent species have significant SNP enrichments with the increasing number of SNP, among which only Eubacterium biforme, Eubacterium hallii and Ruminococcus obeum have relatively high species abundances. Many genes in the microbiomes also presented characteristic SNP distributions between the wet season and the dry season. This implies that the seasonal changes might indirectly impact the mutation patterns for specific species and functions for the gut microbiome of the population that lives in traditional lifestyles through changing the diet in wet and dry seasons, indicating the role of these variants in these species' adaptation to the changing environment and diets.
大多数研究人类肠道微生物组动态的研究都是在居住在城市环境中的人类身上进行的。然而,很少有研究研究过生活在传统生活方式下的人群的肠道微生物组。由于这些人群接触抗生素的机会较少,对自然资源的依赖度较高,因此他们可能是回答人类肠道微生物组进化的更好的研究对象。坦桑尼亚的哈扎狩猎采集者在家庭层面上表现出了很高的肠道微生物组组成的生物多样性和季节性模式,其中一些分类群在一个季节消失,后来又重新出现。已经对这些季节性变化进行了分析,但在基因组水平上核苷酸变化仍未得到探索。因此,微生物群落如何随着基因组水平的季节性变化而变化仍然难以捉摸。
在这项研究中,我们对跨越三个季节的 40 个哈扎粪便宏基因组样本进行了菌株水平的单核苷酸多态性(SNP)分析。在雨季有更多的 SNP 出现,通过 VarScan2 随着 SNP 调用数量的增加,有 8 个优势物种有显著的 SNP 富集,其中只有 3 个物种的相对丰度较高。有 83 个基因在雨季和旱季之间有最多的 SNP 分布。这些基因中的许多来自 Ruminococcus obeum,主要参与代谢途径,包括碳代谢、丙酮酸代谢和糖酵解。
随着 SNP 数量的增加,有 8 个优势物种有显著的 SNP 富集,其中只有 Eubacterium biforme、Eubacterium hallii 和 Ruminococcus obeum 等物种的相对丰度较高。微生物组中的许多基因在雨季和旱季之间也表现出特征性的 SNP 分布。这意味着季节性变化可能通过改变雨季和旱季的饮食,间接影响生活在传统生活方式下的人群肠道微生物组中特定物种和功能的突变模式,表明这些变异在这些物种适应不断变化的环境和饮食方面的作用。