Department of Physics and Astronomy, University of Bologna, 40127, Bologna, BO, Italy.
Department of Experimental, Diagnostic and Specialty Medicine, University of Bologna, 40138, Bologna, BO, Italy.
BMC Bioinformatics. 2021 Feb 9;22(1):60. doi: 10.1186/s12859-020-03780-3.
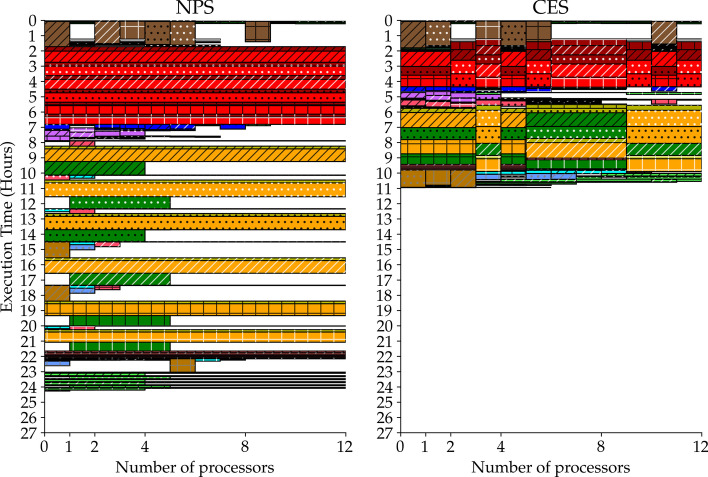
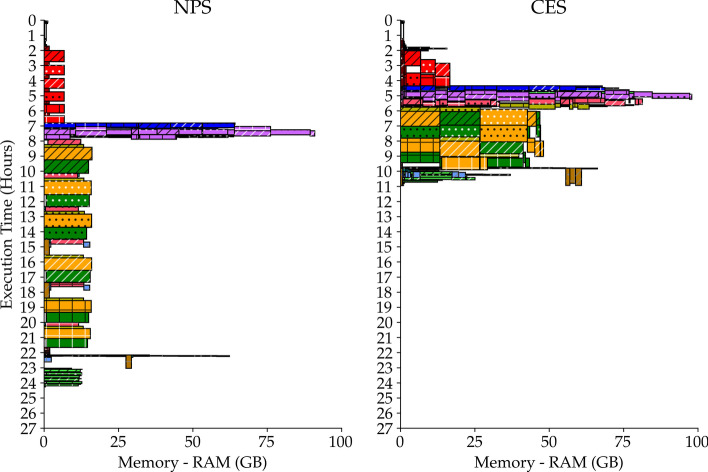
Current high-throughput technologies-i.e. whole genome sequencing, RNA-Seq, ChIP-Seq, etc.-generate huge amounts of data and their usage gets more widespread with each passing year. Complex analysis pipelines involving several computationally-intensive steps have to be applied on an increasing number of samples. Workflow management systems allow parallelization and a more efficient usage of computational power. Nevertheless, this mostly happens by assigning the available cores to a single or few samples' pipeline at a time. We refer to this approach as naive parallel strategy (NPS). Here, we discuss an alternative approach, which we refer to as concurrent execution strategy (CES), which equally distributes the available processors across every sample's pipeline.
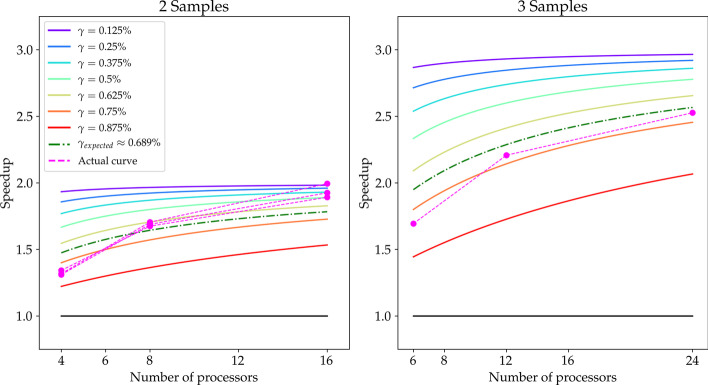
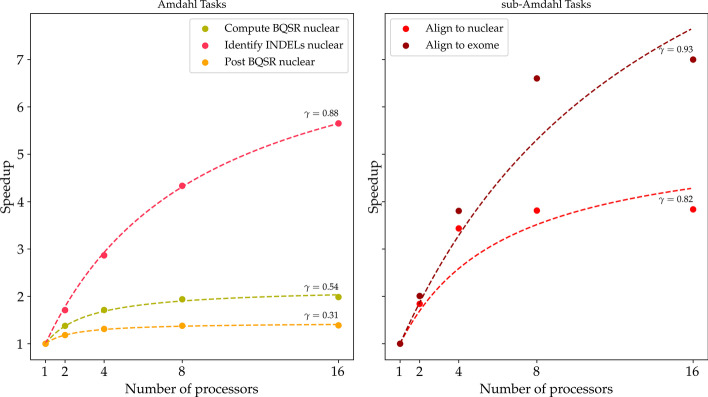
Theoretically, we show that the CES results, under loose conditions, in a substantial speedup, with an ideal gain range spanning from 1 to the number of samples. Also, we observe that the CES yields even faster executions since parallelly computable tasks scale sub-linearly. Practically, we tested both strategies on a whole exome sequencing pipeline applied to three publicly available matched tumour-normal sample pairs of gastrointestinal stromal tumour. The CES achieved speedups in latency up to 2-2.4 compared to the NPS.
Our results hint that if resources distribution is further tailored to fit specific situations, an even greater gain in performance of multiple samples pipelines execution could be achieved. For this to be feasible, a benchmarking of the tools included in the pipeline would be necessary. It is our opinion these benchmarks should be consistently performed by the tools' developers. Finally, these results suggest that concurrent strategies might also lead to energy and cost savings by making feasible the usage of low power machine clusters.
当前的高通量技术——例如全基因组测序、RNA-Seq、ChIP-Seq 等——产生了大量的数据,并且随着时间的推移,它们的使用越来越广泛。涉及多个计算密集型步骤的复杂分析管道必须应用于越来越多的样本。工作流管理系统允许并行化和更有效地利用计算能力。然而,这主要是通过将可用的核心一次分配给一个或几个样本的管道来实现的。我们将这种方法称为朴素并行策略(NPS)。在这里,我们讨论一种替代方法,我们称之为并发执行策略(CES),它将可用的处理器平均分配到每个样本的管道中。
从理论上讲,我们表明,在宽松的条件下,CES 会导致实质性的加速,理想的增益范围从 1 到样本数量。此外,我们观察到,由于并行可计算任务呈次线性扩展,CES 产生的执行速度甚至更快。实际上,我们在应用于三个公开可用的胃肠道间质瘤匹配肿瘤-正常样本对的全外显子测序管道上测试了这两种策略。与 NPS 相比,CES 在延迟方面实现了高达 2-2.4 倍的加速。
我们的结果表明,如果进一步调整资源分配以适应特定情况,那么可以实现多个样本管道执行性能的更大提升。为此,有必要对管道中包含的工具进行基准测试。我们认为,这些基准测试应由工具的开发人员来执行。最后,这些结果表明,通过使低功耗机器集群的使用成为可能,并发策略也可能导致能源和成本节约。