Husev Mykola, Rovenchak Andrij
Department for Theoretical Physics, Ivan Franko National University of Lviv, 12 Drahomanov St, UA-79005 Lviv, Ukraine.
Biosemiotics. 2021;14(2):253-269. doi: 10.1007/s12304-021-09403-5. Epub 2021 Feb 17.
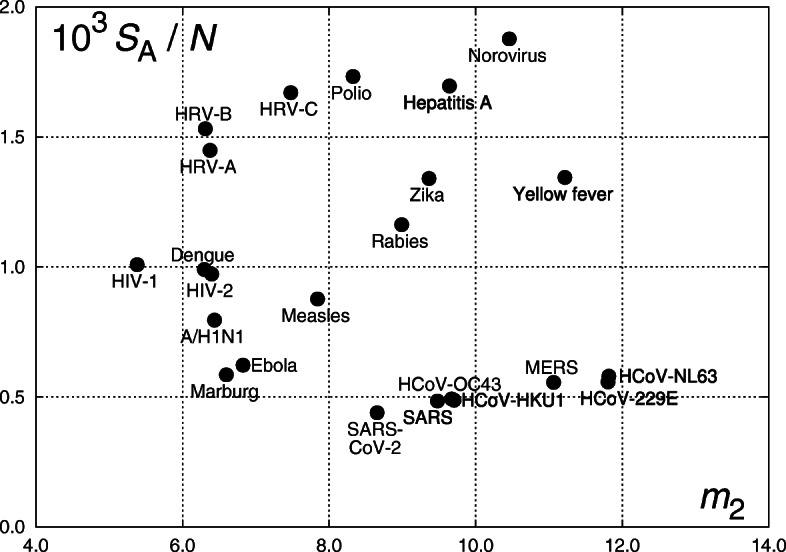
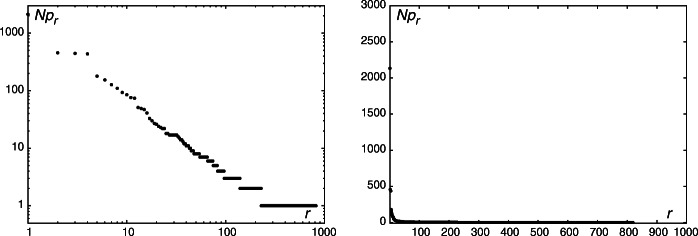
The aim of the study is to analyze viruses using parameters obtained from distributions of nucleotide sequences in the viral RNA. Seeking for the input data homogeneity, we analyze single-stranded RNA viruses only. Two approaches are used to obtain the nucleotide sequences; In the first one, chunks of equal length (four nucleotides) are considered. In the second approach, the whole RNA genome is divided into parts by adenine or the most frequent nucleotide as a "space". Rank-frequency distributions are studied in both cases. The defined nucleotide sequences are signs comparable to a certain extent to syllables or words as seen from the nature of their rank-frequency distributions. Within the first approach, the Pólya and the negative hypergeometric distribution yield the best fit. For the distributions obtained within the second approach, we have calculated a set of parameters, including entropy, mean sequence length, and its dispersion. The calculated parameters became the basis for the classification of viruses. We observed that proximity of viruses on planes spanned on various pairs of parameters corresponds to related species. In certain cases, such a proximity is observed for unrelated species as well calling thus for the expansion of the set of parameters used in the classification. We also observed that the fifth most frequent nucleotide sequences obtained within the second approach are of different nature in case of human coronaviruses (different nucleotides for MERS, SARS-CoV, and SARS-CoV-2 versus identical nucleotides for four other coronaviruses). We expect that our findings will be useful as a supplementary tool in the classification of diseases caused by RNA viruses with respect to severity and contagiousness.
本研究的目的是利用从病毒RNA核苷酸序列分布中获得的参数来分析病毒。为了寻求输入数据的同质性,我们仅分析单链RNA病毒。使用两种方法来获取核苷酸序列;在第一种方法中,考虑等长的片段(四个核苷酸)。在第二种方法中,整个RNA基因组以腺嘌呤或最常见的核苷酸作为“间隔”被分成多个部分。在这两种情况下都研究了秩频分布。从其秩频分布的性质来看,所定义的核苷酸序列在一定程度上类似于音节或单词。在第一种方法中,波利亚分布和负超几何分布拟合效果最佳。对于在第二种方法中获得的分布,我们计算了一组参数,包括熵、平均序列长度及其离散度。计算出的参数成为病毒分类的基础。我们观察到,在由各种参数对构成的平面上病毒的接近程度对应于相关物种。在某些情况下,不相关物种之间也观察到这种接近程度,因此需要扩展分类中使用的参数集。我们还观察到,在第二种方法中获得的第五个最常见的核苷酸序列在人类冠状病毒的情况下具有不同的性质(中东呼吸综合征冠状病毒、严重急性呼吸综合征冠状病毒和严重急性呼吸综合征冠状病毒2的核苷酸不同,而其他四种冠状病毒的核苷酸相同)。我们期望我们的发现将作为一种补充工具,用于根据严重程度和传染性对由RNA病毒引起的疾病进行分类。