Ren Jie, Song Kai, Deng Chao, Ahlgren Nathan A, Fuhrman Jed A, Li Yi, Xie Xiaohui, Poplin Ryan, Sun Fengzhu
Quantitative and Computational Biology Program, University of Southern California, Los Angeles, CA 90089, USA.
School of Mathematics and Statistics, Qingdao University, Qingdao 266071, China.
Quant Biol. 2020 Mar;8(1):64-77. doi: 10.1007/s40484-019-0187-4.
The recent development of metagenomic sequencing makes it possible to massively sequence microbial genomes including viral genomes without the need for laboratory culture. Existing reference-based and gene homology-based methods are not efficient in identifying unknown viruses or short viral sequences from metagenomic data.
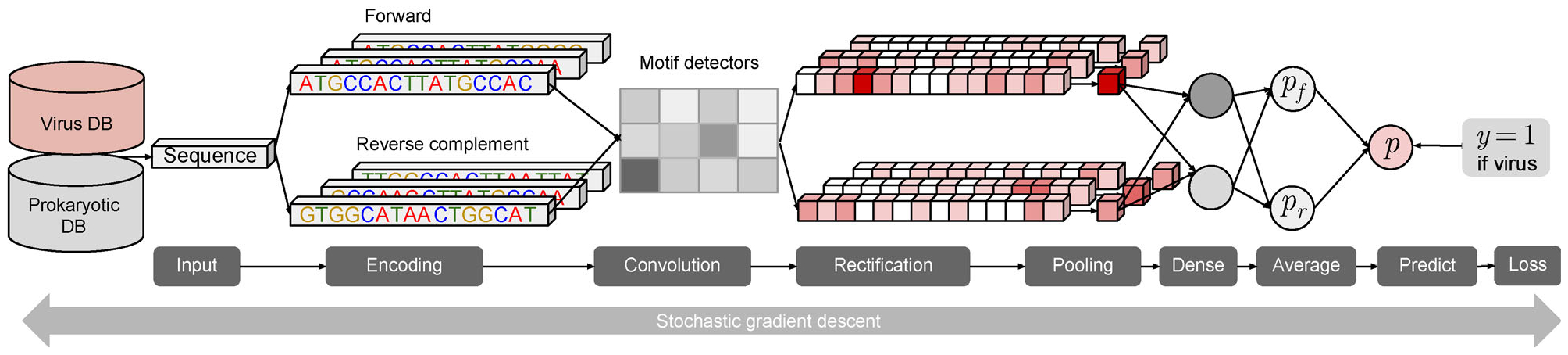
Here we developed a reference-free and alignment-free machine learning method, DeepVirFinder, for identifying viral sequences in metagenomic data using deep learning.
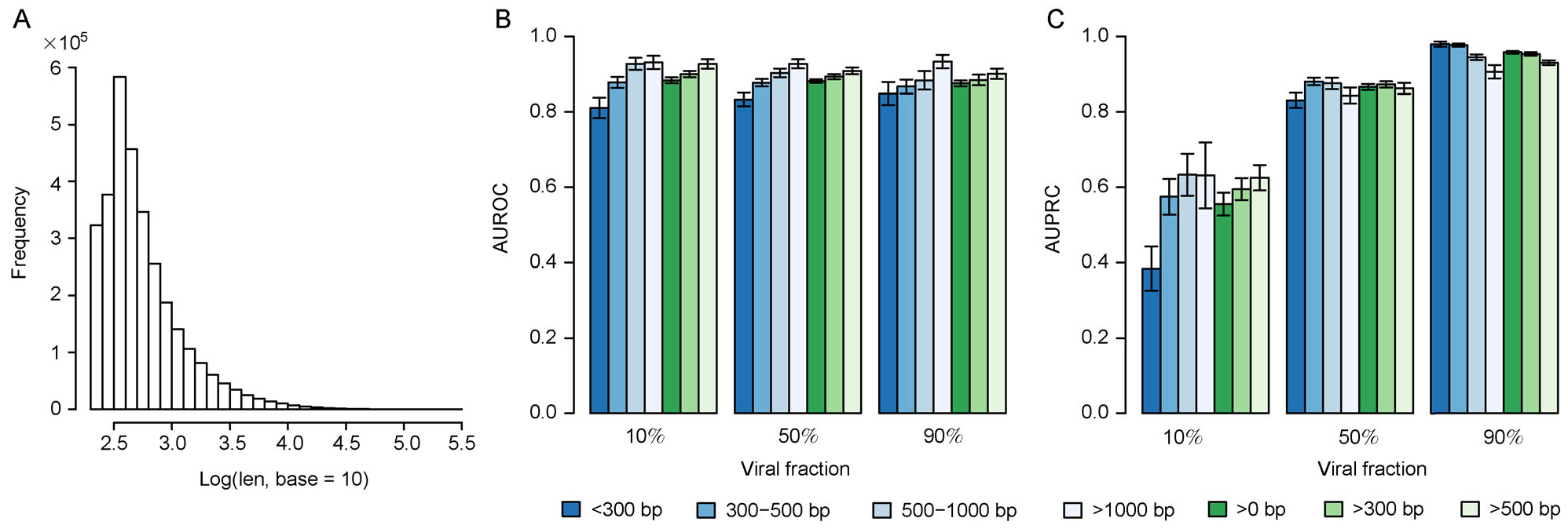
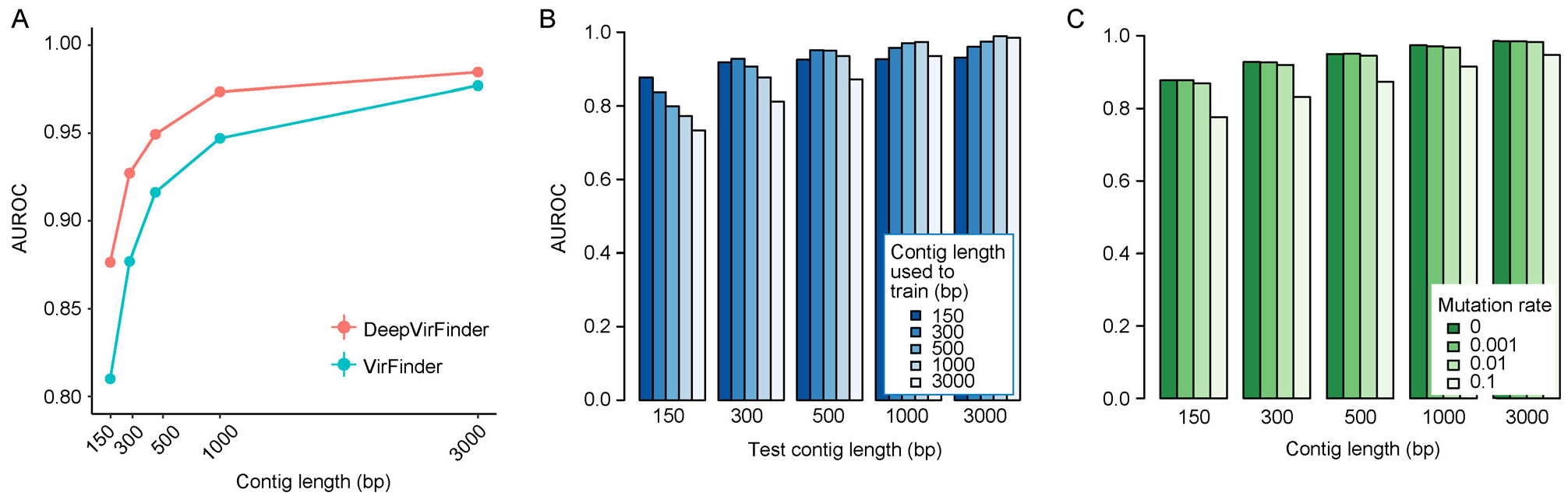
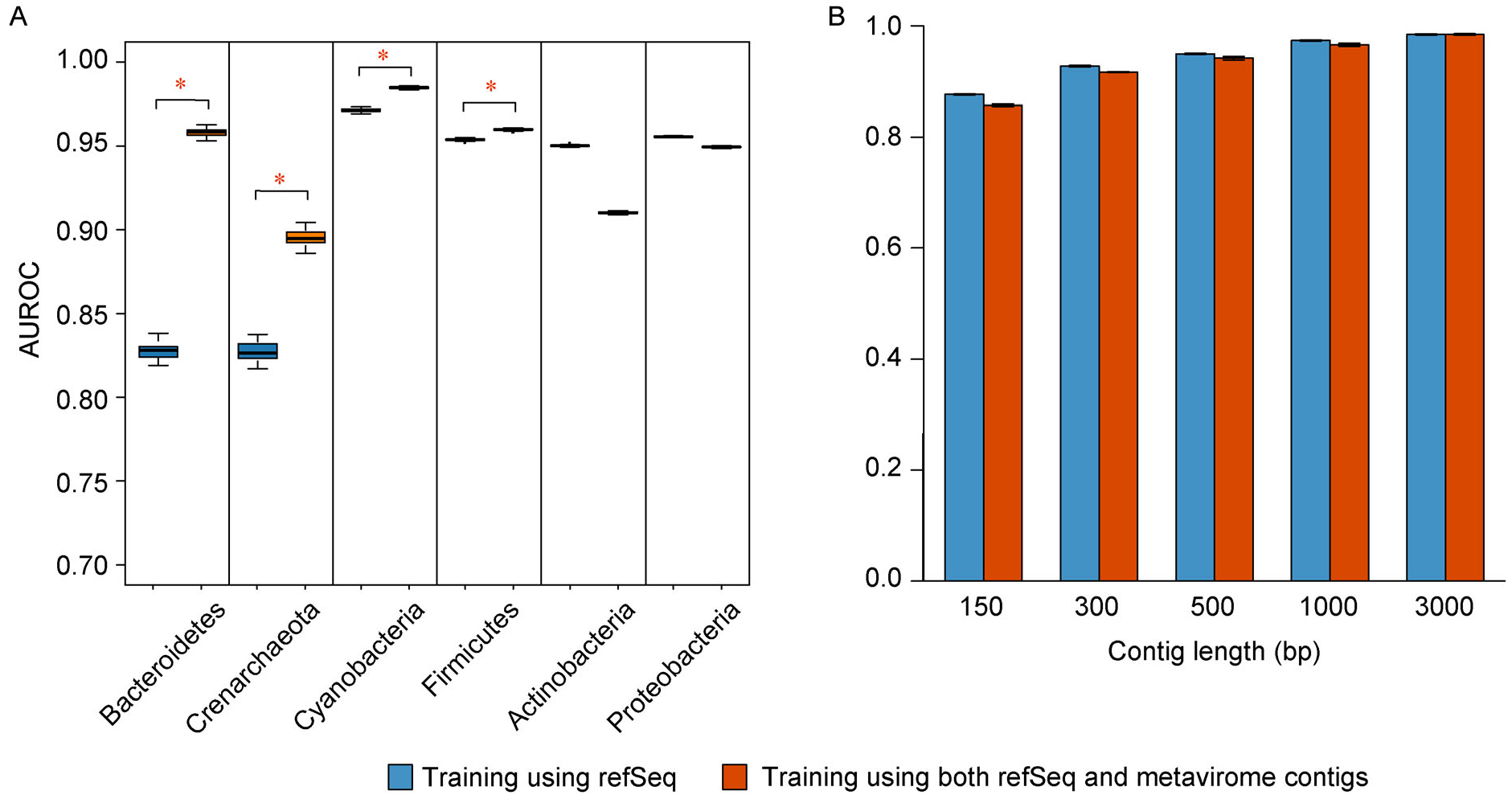
Trained based on sequences from viral RefSeq discovered before May 2015, and evaluated on those discovered after that date, DeepVirFinder outperformed the state-of-the-art method VirFinder at all contig lengths, achieving AUROC 0.93, 0.95, 0.97, and 0.98 for 300, 500, 1000, and 3000 bp sequences respectively. Enlarging the training data with additional millions of purified viral sequences from metavirome samples further improved the accuracy for identifying virus groups that are under-represented. Applying DeepVirFinder to real human gut metagenomic samples, we identified 51,138 viral sequences belonging to 175 bins in patients with colorectal carcinoma (CRC). Ten bins were found associated with the cancer status, suggesting viruses may play important roles in CRC.
Powered by deep learning and high throughput sequencing metagenomic data, DeepVirFinder significantly improved the accuracy of viral identification and will assist the study of viruses in the era of metagenomics.
宏基因组测序技术的最新发展使得对包括病毒基因组在内的微生物基因组进行大规模测序成为可能,而无需实验室培养。现有的基于参考序列和基因同源性的方法在从宏基因组数据中识别未知病毒或短病毒序列方面效率不高。
在此,我们开发了一种无参考序列和无比对的机器学习方法DeepVirFinder,用于利用深度学习识别宏基因组数据中的病毒序列。
基于2015年5月之前发现的病毒RefSeq序列进行训练,并在该日期之后发现的序列上进行评估,DeepVirFinder在所有重叠群长度上均优于当前最先进的方法VirFinder,对于300、500、1000和3000bp序列,分别实现了0.93、0.95、0.97和0.98的曲线下面积(AUROC)。用来自宏病毒组样本的数百万条纯化病毒序列扩大训练数据,进一步提高了识别代表性不足的病毒组的准确性。将DeepVirFinder应用于真实的人类肠道宏基因组样本,我们在结直肠癌(CRC)患者中鉴定出属于175个分类单元的51,138条病毒序列。发现有10个分类单元与癌症状态相关,这表明病毒可能在CRC中起重要作用。
借助深度学习和高通量测序宏基因组数据,DeepVirFinder显著提高了病毒识别的准确性,并将有助于宏基因组学时代的病毒研究。