School of BioSciences, Bio21 Institute, The University of Melbourne, Melbourne, Australia.
Bioinformatics Division, Walter and Eliza Hall Institute, Melbourne, Australia.
PLoS Genet. 2021 Feb 25;17(2):e1009269. doi: 10.1371/journal.pgen.1009269. eCollection 2021 Feb.
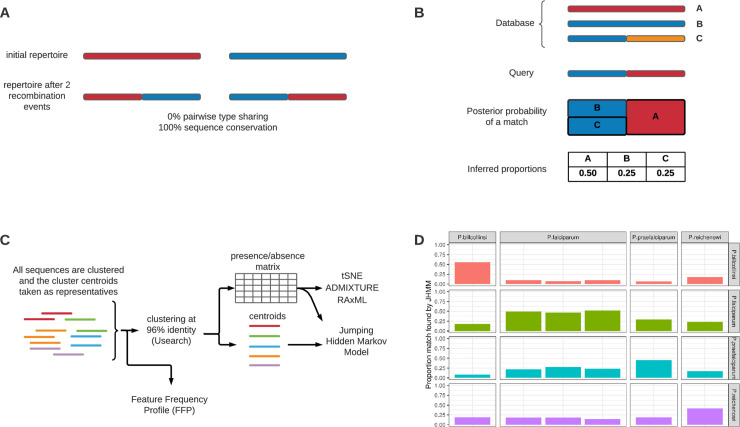
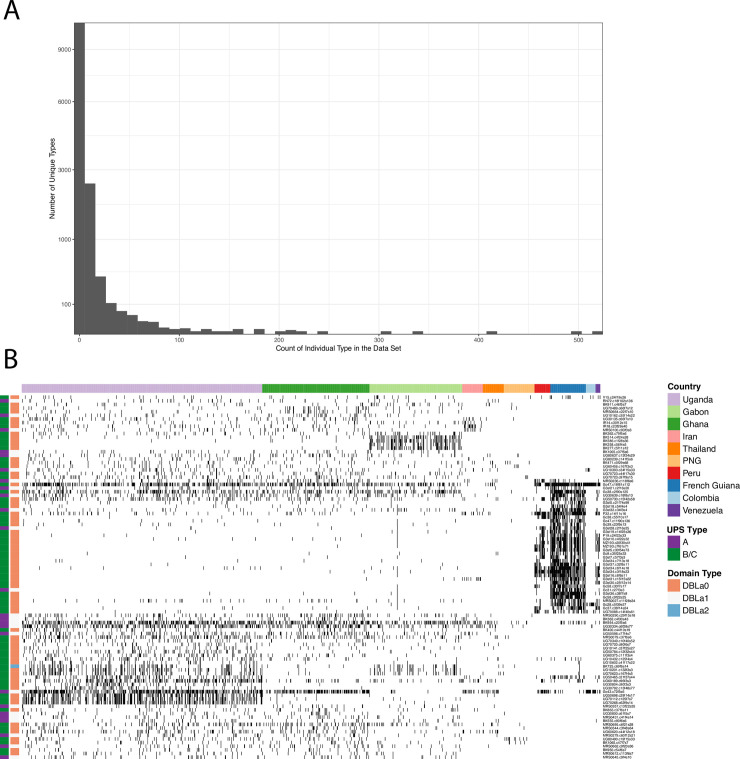
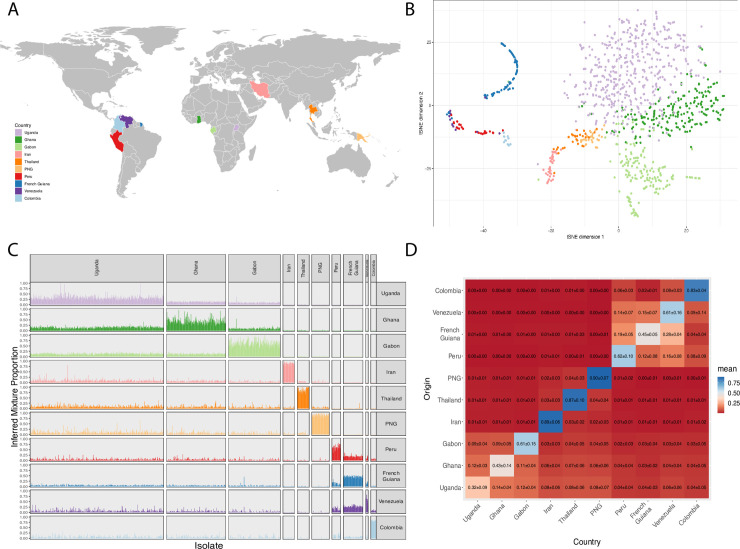
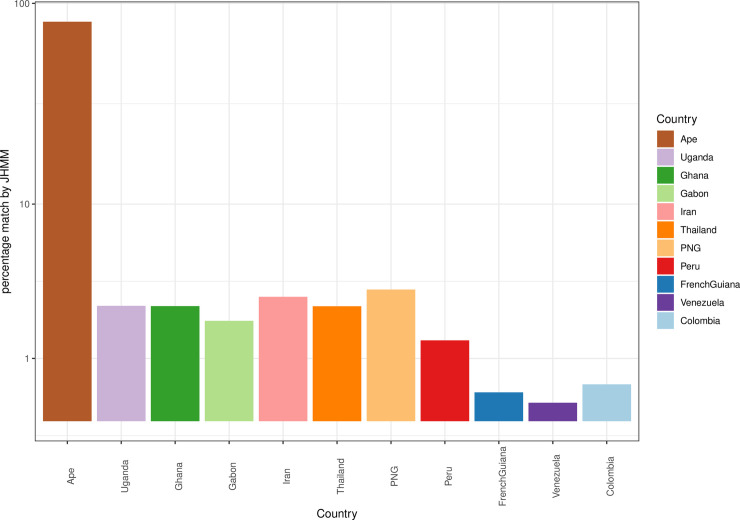
Malaria remains a major public health problem in many countries. Unlike influenza and HIV, where diversity in immunodominant surface antigens is understood geographically to inform disease surveillance, relatively little is known about the global population structure of PfEMP1, the major variant surface antigen of the malaria parasite Plasmodium falciparum. The complexity of the var multigene family that encodes PfEMP1 and that diversifies by recombination, has so far precluded its use in malaria surveillance. Recent studies have demonstrated that cost-effective deep sequencing of the region of var genes encoding the PfEMP1 DBLα domain and subsequent classification of within host sequences at 96% identity to define unique DBLα types, can reveal structure and strain dynamics within countries. However, to date there has not been a comprehensive comparison of these DBLα types between countries. By leveraging a bioinformatic approach (jumping hidden Markov model) designed specifically for the analysis of recombination within var genes and applying it to a dataset of DBLα types from 10 countries, we are able to describe population structure of DBLα types at the global scale. The sensitivity of the approach allows for the comparison of the global dataset to ape samples of Plasmodium Laverania species. Our analyses show that the evolution of the parasite population emerging out of Africa underlies current patterns of DBLα type diversity. Most importantly, we can distinguish geographic population structure within Africa between Gabon and Ghana in West Africa and Uganda in East Africa. Our evolutionary findings have translational implications in the context of globalization. Firstly, DBLα type diversity can provide a simple diagnostic framework for geographic surveillance of the rapidly evolving transmission dynamics of P. falciparum. It can also inform efforts to understand the presence or absence of global, regional and local population immunity to major surface antigen variants. Additionally, we identify a number of highly conserved DBLα types that are present globally that may be of biological significance and warrant further characterization.
疟疾仍然是许多国家的主要公共卫生问题。与流感和艾滋病毒不同,流感和艾滋病毒的免疫显性表面抗原的多样性在地理上被理解为疾病监测的信息,而关于疟原虫恶性疟原虫主要变异表面抗原 PfEMP1 的全球种群结构,人们知之甚少。PfEMP1 由 var 多基因家族编码,该基因家族通过重组多样化,其复杂性使得它迄今为止无法用于疟疾监测。最近的研究表明,对编码 PfEMP1 DBLα 结构域的 var 基因区域进行经济有效的深度测序,随后对 96%同一性的宿主内序列进行分类,以定义独特的 DBLα 类型,可以揭示国家内部的结构和菌株动态。然而,迄今为止,还没有对这些 DBLα 类型在国家之间进行全面比较。通过利用专门针对 var 基因内重组分析而设计的生物信息学方法(跳跃隐马尔可夫模型),并将其应用于来自 10 个国家的 DBLα 类型数据集,我们能够描述全球范围内 DBLα 类型的种群结构。该方法的敏感性允许将全球数据集与 Plasmodium Laverania 物种的 ape 样本进行比较。我们的分析表明,在非洲出现的寄生虫种群的进化是当前 DBLα 类型多样性模式的基础。最重要的是,我们可以区分非洲内部的地理种群结构,包括西非的加蓬和加纳以及东非的乌干达。我们的进化发现对全球化背景下具有转化意义。首先,DBLα 类型多样性可以为 Pfalciparum 快速演变的传播动态的地理监测提供一个简单的诊断框架。它还可以为了解全球、区域和局部人群对主要表面抗原变异体的存在或不存在提供信息。此外,我们确定了一些在全球范围内存在的高度保守的 DBLα 类型,它们可能具有生物学意义,值得进一步研究。