Waminal Nomar Espinosa, Pellerin Remnyl Joyce, Kang Sang-Ho, Kim Hyun Hee
Department of Chemistry and Life Science, BioScience Institute, Sahmyook University, Seoul, South Korea.
Genomics Division, National Institute of Agricultural Sciences, Rural Development Administration, Jeonju, South Korea.
Front Plant Sci. 2021 Feb 10;12:629898. doi: 10.3389/fpls.2021.629898. eCollection 2021.
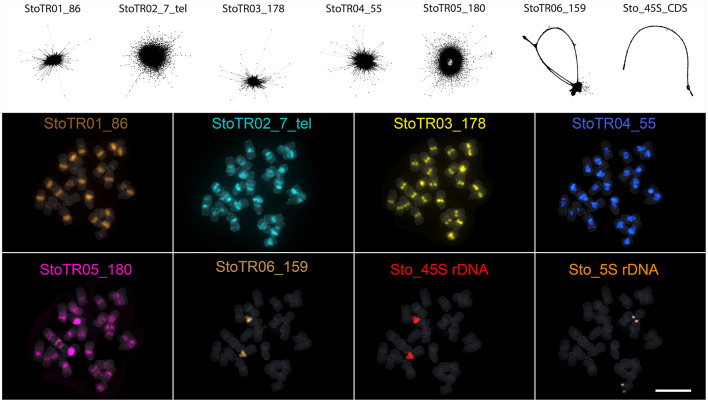
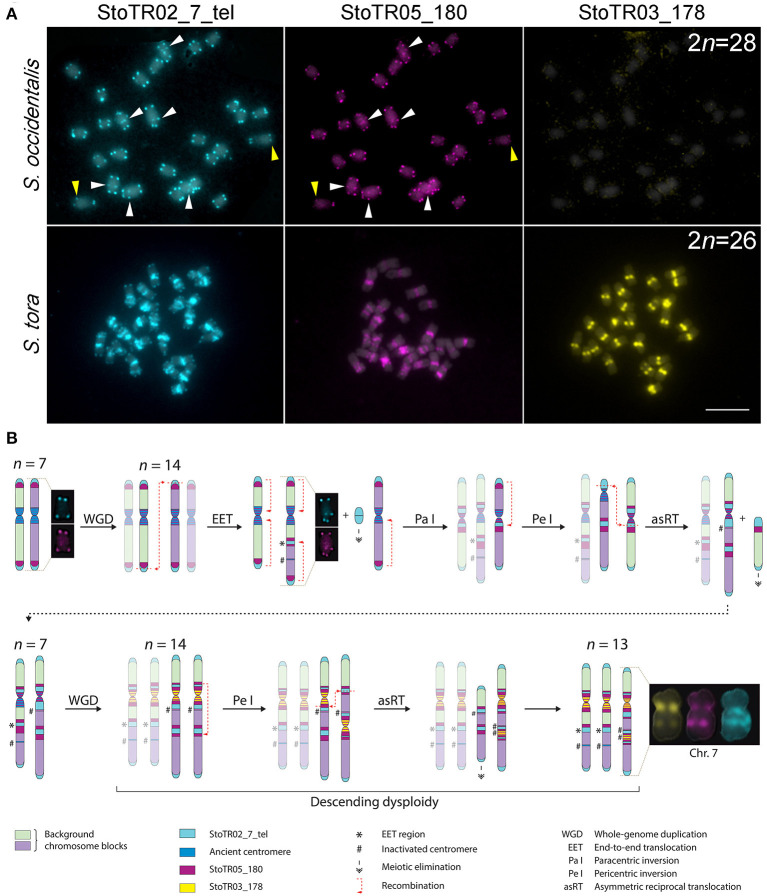
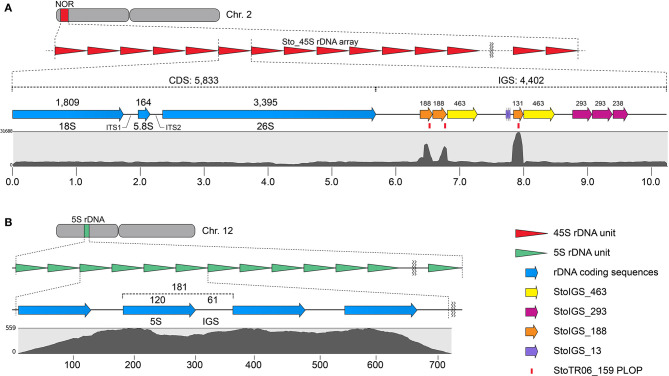
Tandem repeats can occupy a large portion of plant genomes and can either cause or result from chromosomal rearrangements, which are important drivers of dysploidy-mediated karyotype evolution and speciation. To understand the contribution of tandem repeats in shaping the extant dysploid karyotype, we analyzed the composition and abundance of tandem repeats in the genome and compared the chromosomal distribution of these repeats between and a closely related euploid, . Using a read clustering algorithm, we identified the major tandem repeats and visualized their chromosomal distribution by fluorescence hybridization. We identified eight independent repeats covering ~85 Mb or ~12% of the genome. The unit lengths and copy numbers had ranges of 7-5,833 bp and 325-2.89 × 10, respectively. Three short duplicated sequences were found in the 45S rDNA intergenic spacer, one of which was also detected at an extra-NOR locus. The canonical plant telomeric repeat (TTTAGGG) was also detected as very intense signals in numerous pericentromeric and interstitial loci. StoTR05_180, which showed subtelomeric distribution in , was predominantly pericentromeric in . The unusual chromosomal distribution of tandem repeats in not only enabled easy identification of individual chromosomes but also revealed the massive chromosomal rearrangements that have likely played important roles in shaping its dysploid karyotype.
串联重复序列可占据植物基因组的很大一部分,并且既可能导致染色体重排,也可能由染色体重排产生,而染色体重排是倍性异常介导的核型进化和物种形成的重要驱动因素。为了了解串联重复序列在塑造现存倍性异常核型中的作用,我们分析了基因组中串联重复序列的组成和丰度,并比较了这些重复序列在[物种名称1]及其近缘整倍体[物种名称2]之间的染色体分布。我们使用一种 reads 聚类算法鉴定了主要的串联重复序列,并通过荧光原位杂交可视化了它们的染色体分布。我们鉴定出八个独立的重复序列,覆盖了约 85 Mb 或基因组的约 12%。其单位长度和拷贝数范围分别为 7 - 5833 bp 和 325 - 2.89×10⁴。在 45S rDNA 基因间隔区发现了三个短重复序列,其中一个也在核仁组织区以外的位点被检测到。典型的植物端粒重复序列(TTTAGGG)在许多着丝粒周围和居间位点也被检测为非常强烈的信号。在[物种名称1]中显示为亚端粒分布的 StoTR05_180,在[物种名称2]中主要位于着丝粒周围。[物种名称1]中串联重复序列异常的染色体分布不仅便于识别单个染色体,还揭示了可能在塑造其倍性异常核型中发挥重要作用的大规模染色体重排。