Graduate Program in Biomolecular Structure and Design, University of Washington, Seattle, Washington, United States of America.
PLoS One. 2011;6(7):e22060. doi: 10.1371/journal.pone.0022060. Epub 2011 Jul 29.
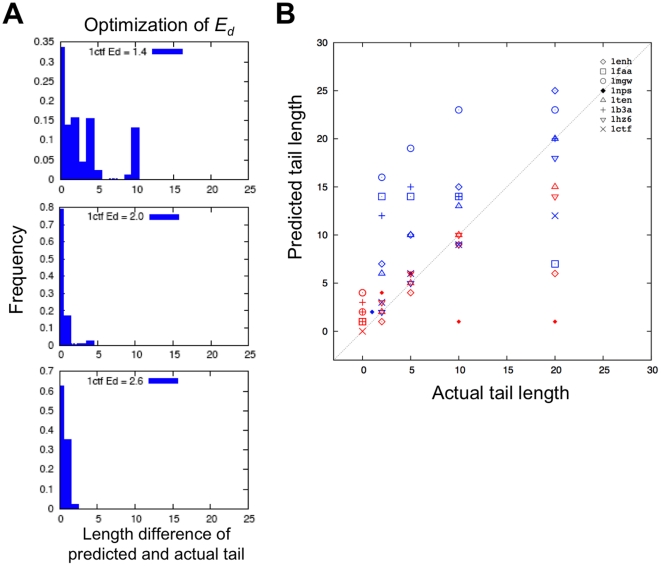
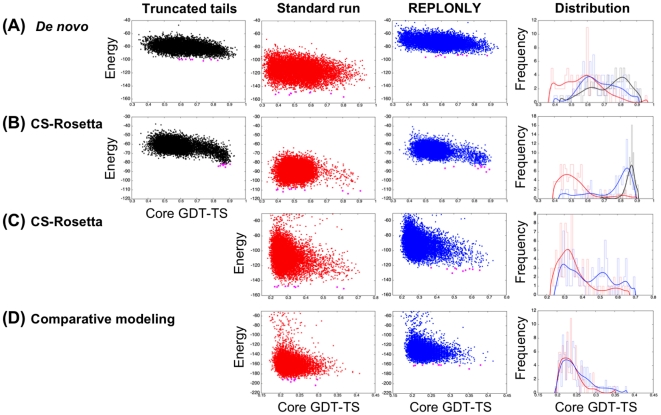
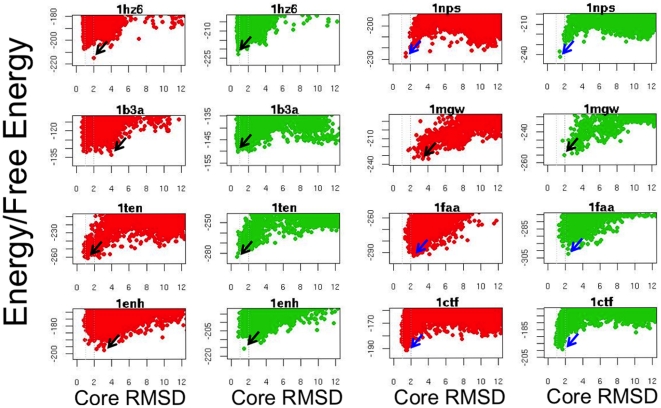
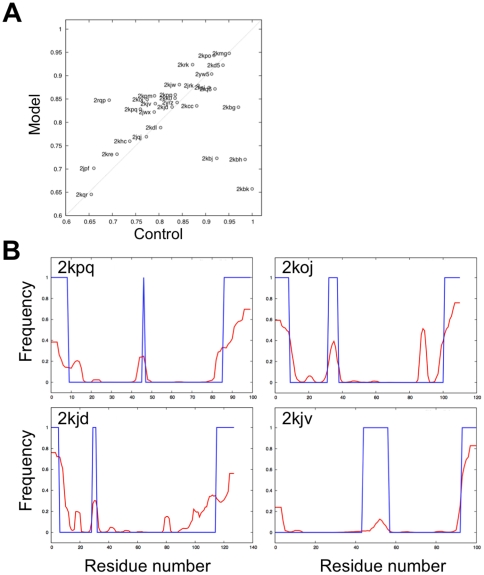
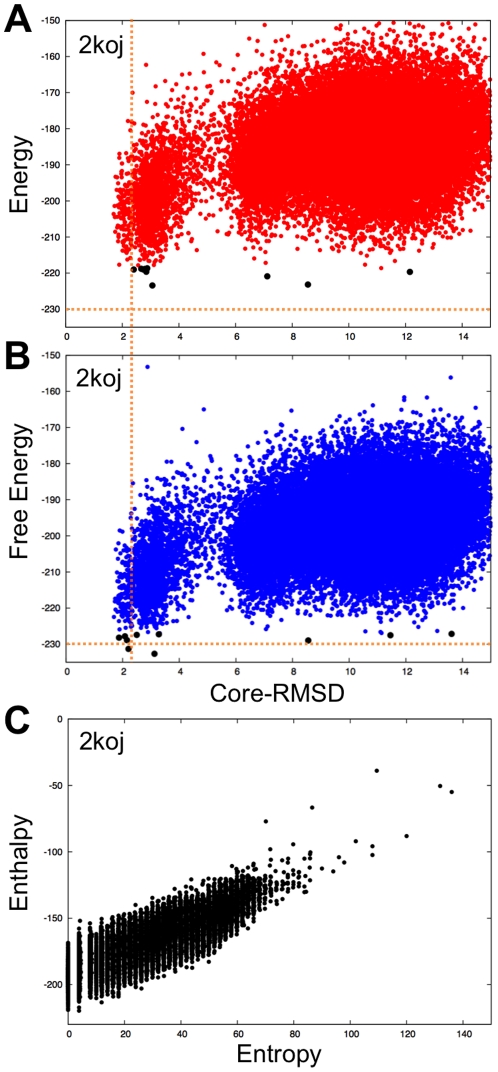
Protein structure prediction methods such as Rosetta search for the lowest energy conformation of the polypeptide chain. However, the experimentally observed native state is at a minimum of the free energy, rather than the energy. The neglect of the missing configurational entropy contribution to the free energy can be partially justified by the assumption that the entropies of alternative folded states, while very much less than unfolded states, are not too different from one another, and hence can be to a first approximation neglected when searching for the lowest free energy state. The shortcomings of current structure prediction methods may be due in part to the breakdown of this assumption. Particularly problematic are proteins with significant disordered regions which do not populate single low energy conformations even in the native state. We describe two approaches within the Rosetta structure modeling methodology for treating such regions. The first does not require advance knowledge of the regions likely to be disordered; instead these are identified by minimizing a simple free energy function used previously to model protein folding landscapes and transition states. In this model, residues can be either completely ordered or completely disordered; they are considered disordered if the gain in entropy outweighs the loss of favorable energetic interactions with the rest of the protein chain. The second approach requires identification in advance of the disordered regions either from sequence alone using for example the DISOPRED server or from experimental data such as NMR chemical shifts. During Rosetta structure prediction calculations the disordered regions make only unfavorable repulsive contributions to the total energy. We find that the second approach has greater practical utility and illustrate this with examples from de novo structure prediction, NMR structure calculation, and comparative modeling.
蛋白质结构预测方法,如 Rosetta 搜索,寻找多肽链的最低能量构象。然而,实验观察到的天然状态是自由能的最小值,而不是能量的最小值。自由能中忽略缺失的构象熵贡献可以部分地通过假设来证明,即替代折叠状态的熵,虽然远小于未折叠状态,但彼此之间没有太大区别,因此在搜索最低自由能状态时可以近似忽略。当前结构预测方法的缺点可能部分归因于这一假设的失效。对于那些具有显著无序区域的蛋白质来说,问题尤其严重,即使在天然状态下,它们也不会形成单一的低能量构象。我们在 Rosetta 结构建模方法中描述了两种处理这些区域的方法。第一种方法不需要预先了解可能无序的区域;相反,这些区域是通过最小化一个简单的自由能函数来识别的,该函数以前曾用于模拟蛋白质折叠景观和过渡态。在这个模型中,残基可以是完全有序的,也可以是完全无序的;如果熵的增加超过了与蛋白质链其余部分有利的能量相互作用的损失,那么它们就被认为是无序的。第二种方法需要预先识别无序区域,要么是从序列本身,例如使用 DISOPRED 服务器,要么是从实验数据,如 NMR 化学位移。在 Rosetta 结构预测计算中,无序区域只对总能量产生不利的排斥贡献。我们发现第二种方法具有更大的实际效用,并通过从头预测结构、NMR 结构计算和比较建模的例子来说明这一点。