Department of Biostatistics and Bioinformatics, Division of Integrative Genomics, Duke University Medical School, Durham, North Carolina 27710, USA.
Center for Genomic and Computational Biology, Duke University Medical School, Durham, North Carolina 27710, USA.
Genome Res. 2021 May;31(5):877-889. doi: 10.1101/gr.269209.120. Epub 2021 Mar 15.
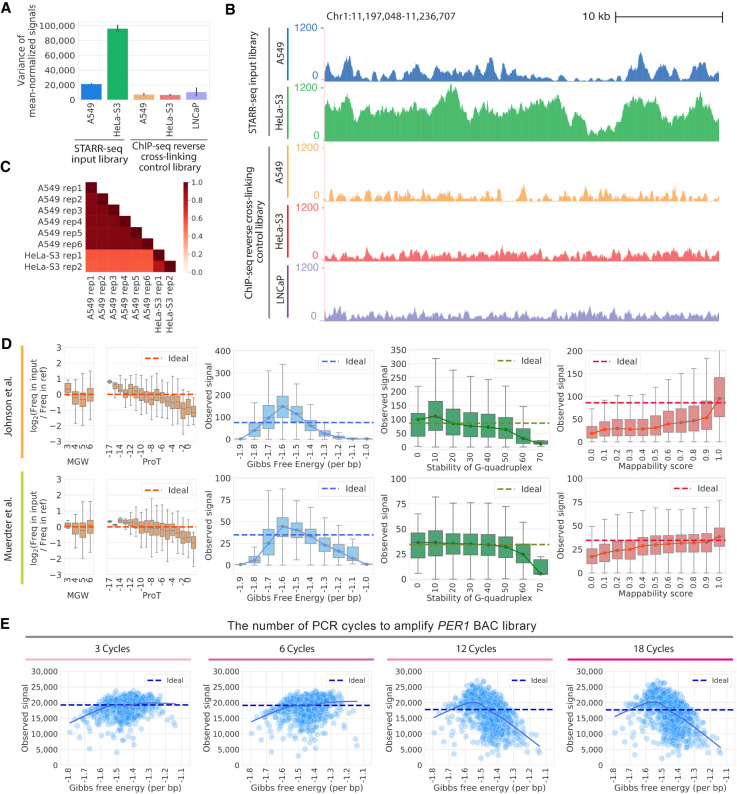
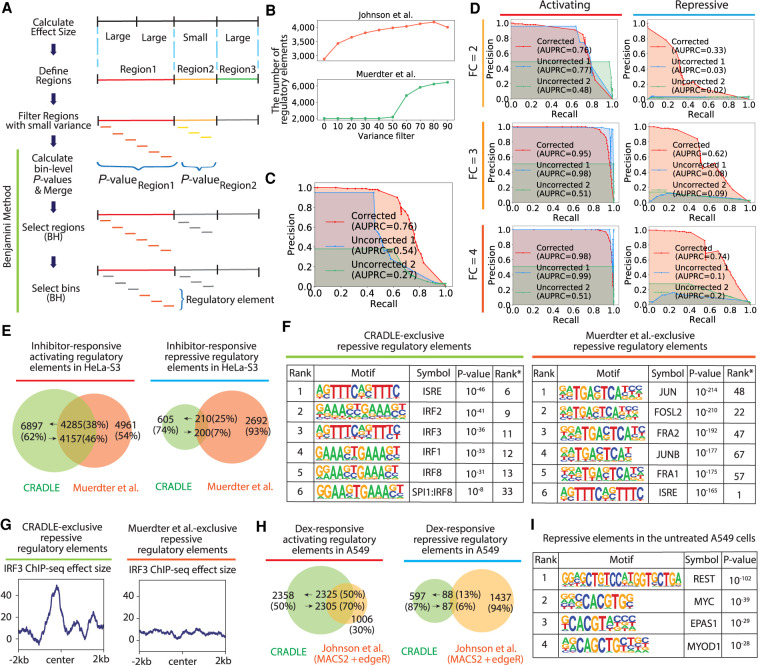
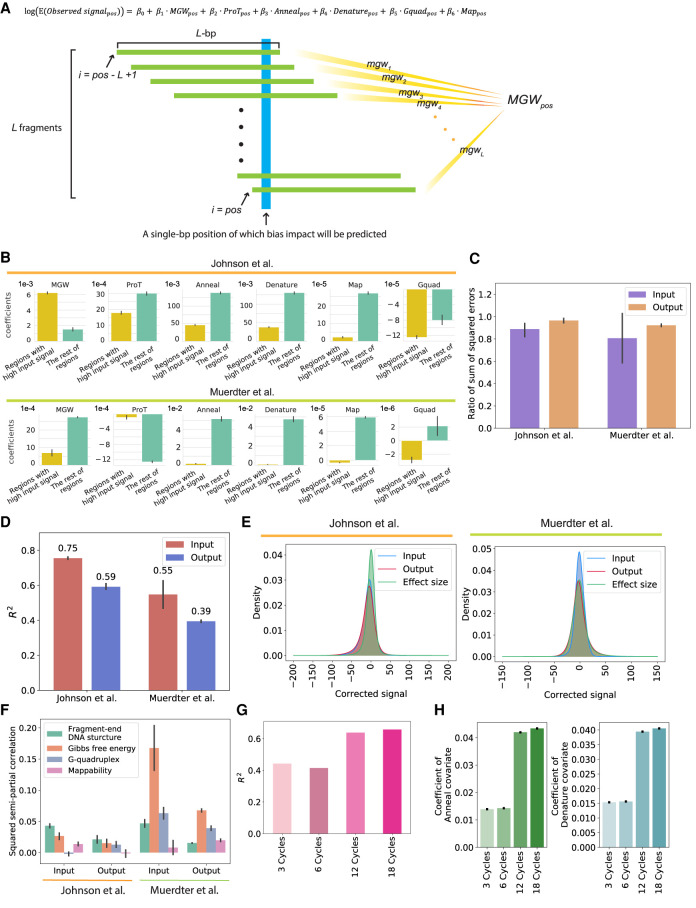
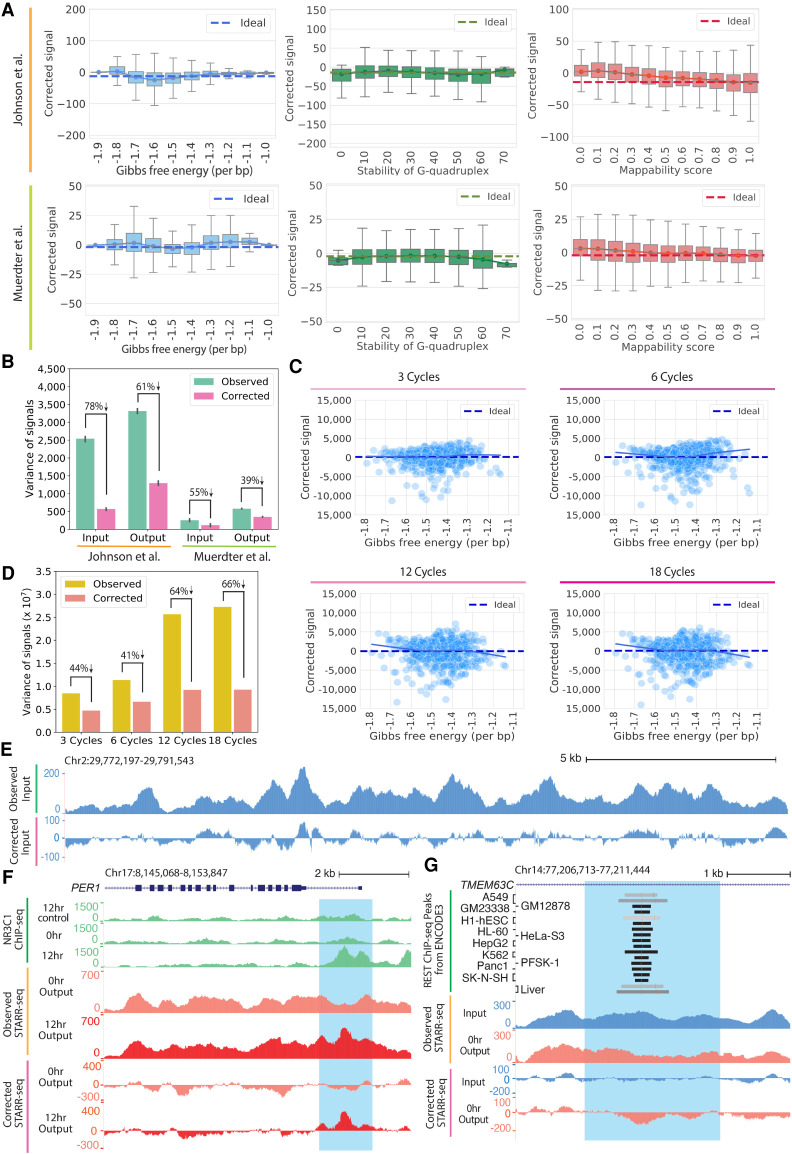
High-throughput reporter assays such as self-transcribing active regulatory region sequencing (STARR-seq) have made it possible to measure regulatory element activity across the entire human genome at once. The resulting data, however, present substantial analytical challenges. Here, we identify technical biases that explain most of the variance in STARR-seq data. We then develop a statistical model to correct those biases and to improve detection of regulatory elements. This approach substantially improves precision and recall over current methods, improves detection of both activating and repressive regulatory elements, and controls for false discoveries despite strong local correlations in signal.
高通量报告基因检测,如自转录活性调控区测序(STARR-seq),使得在全基因组范围内同时测量调控元件活性成为可能。然而,由此产生的数据提出了重大的分析挑战。在这里,我们确定了技术偏差,这些偏差解释了 STARR-seq 数据的大部分方差。然后,我们开发了一个统计模型来纠正这些偏差,并提高调控元件的检测能力。与当前方法相比,这种方法显著提高了检测的精确性和召回率,提高了激活和抑制性调控元件的检测能力,并且在信号存在强烈局部相关性的情况下控制了假阳性发现。