Laboratory of Computational Systems Biotechnology (LCSB), Swiss Federal Institute of Technology (EPFL), 1015, Lausanne, Switzerland.
BMC Bioinformatics. 2021 Mar 20;22(1):134. doi: 10.1186/s12859-021-04066-y.
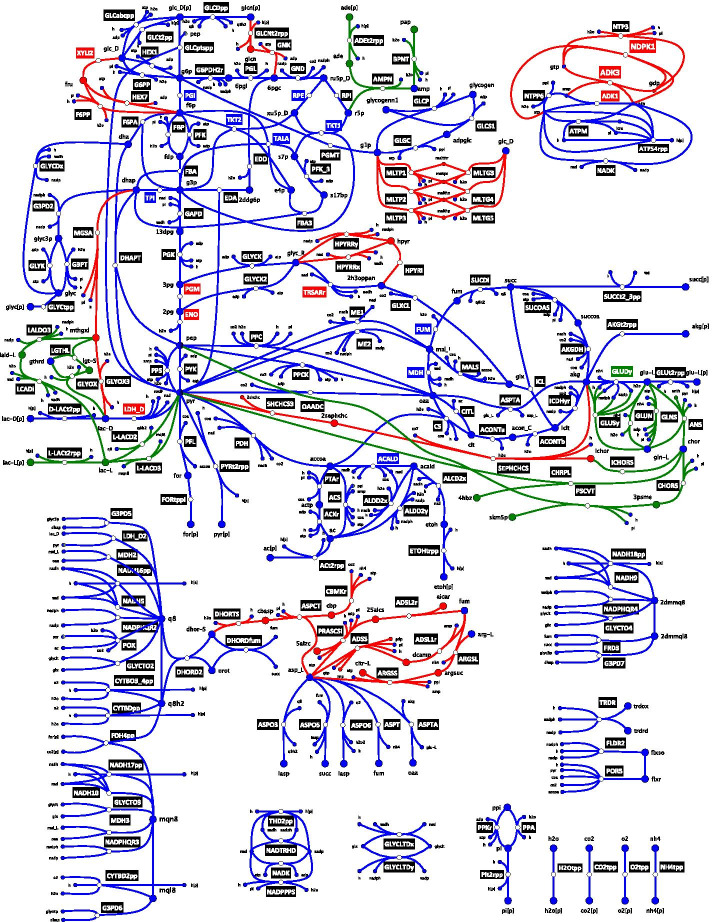
Significant efforts have been made in building large-scale kinetic models of cellular metabolism in the past two decades. However, most kinetic models published to date, remain focused around central carbon pathways or are built around ad hoc reduced models without clear justification on their derivation and usage. Systematic algorithms exist for reducing genome-scale metabolic reconstructions to build thermodynamically feasible and consistently reduced stoichiometric models. However, it is important to study how network complexity affects conclusions derived from large-scale kinetic models built around consistently reduced models before we can apply them to study biological systems.
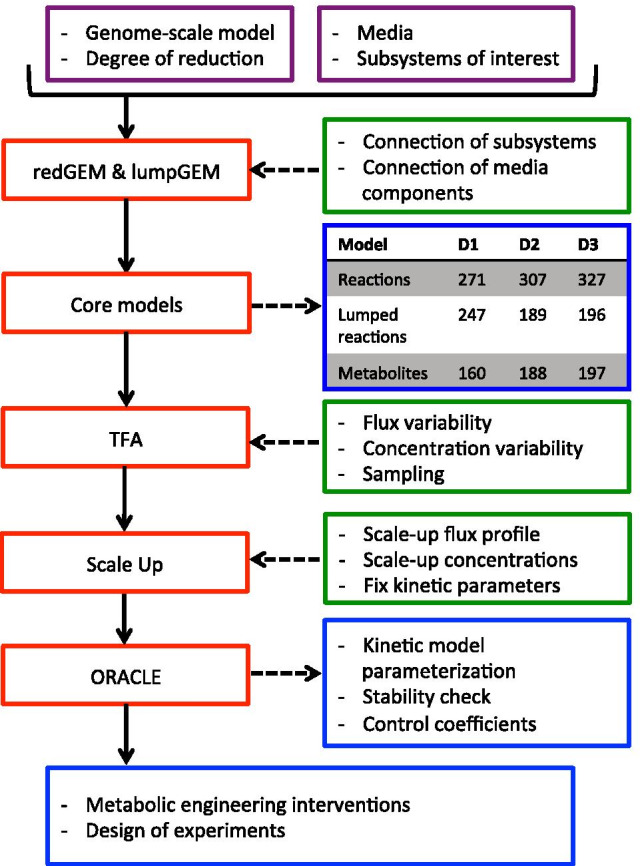
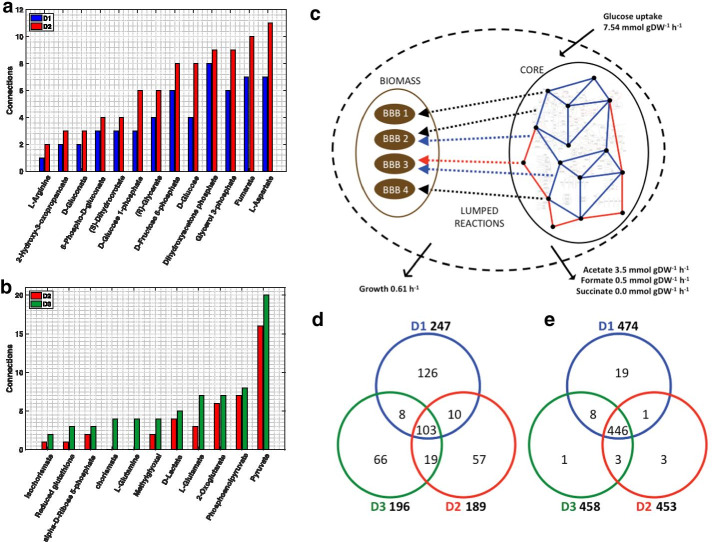
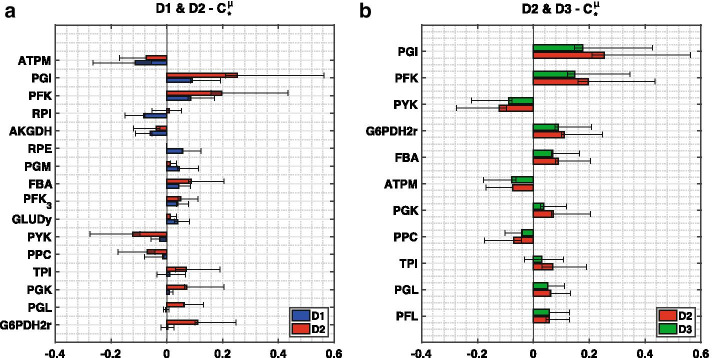
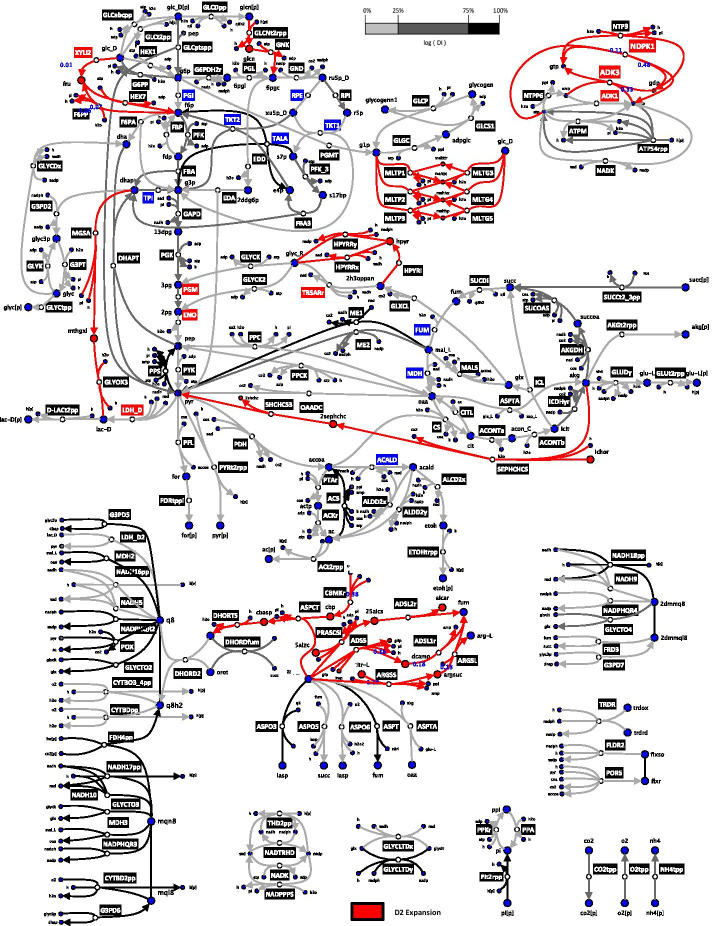
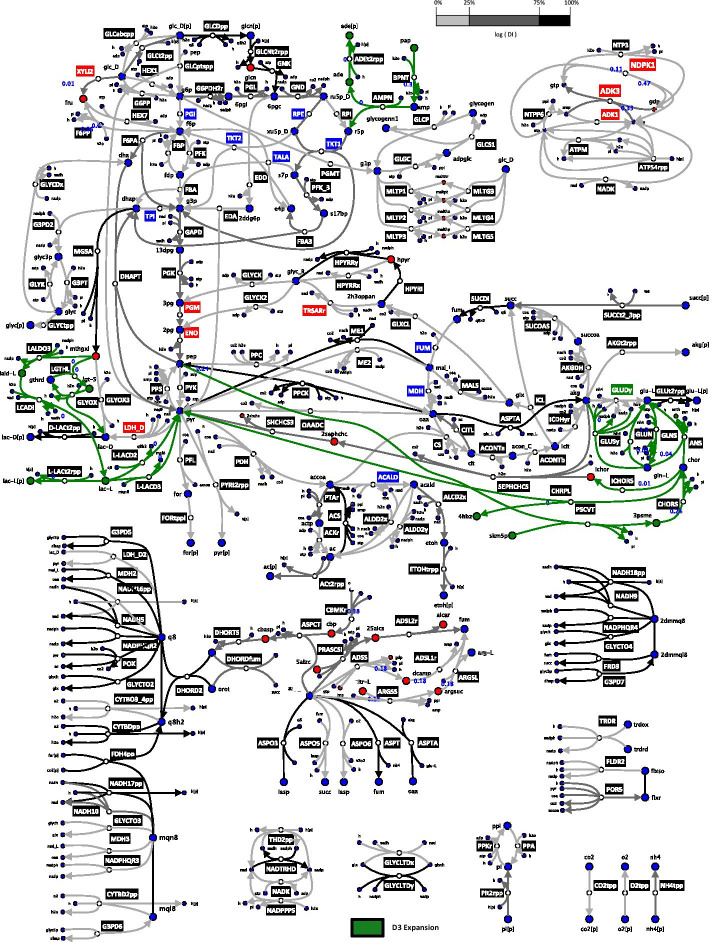
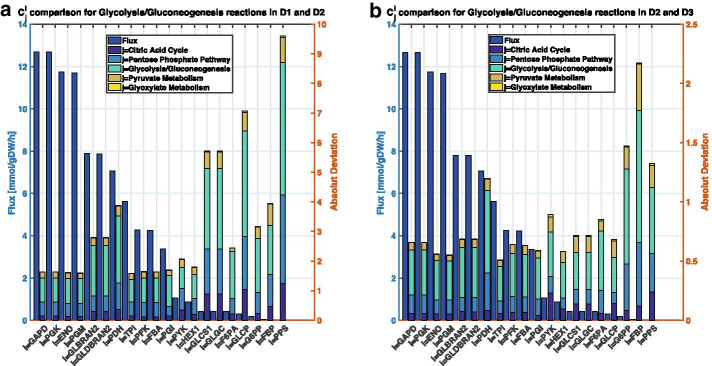
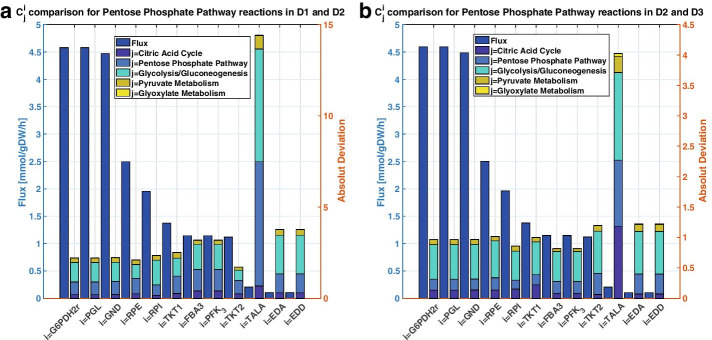
We reduced the iJO1366 Escherichia Coli genome-scale metabolic reconstruction systematically to build three stoichiometric models of different size. Since the reduced models are expansions around the core subsystems for which the reduction was performed, the models are nested. We present a method for scaling up the flux profile and the concentration vector reference steady-states from the smallest model to the larger ones, whilst preserving maximum equivalency. Populations of kinetic models, preserving similarity in kinetic parameters, were built around the reference steady-states and their metabolic sensitivity coefficients (MSCs) were computed. The MSCs were sensitive to the model complexity. We proposed a metric for measuring the sensitivity of MSCs to these structural changes.
We proposed for the first time a workflow for scaling up the size of kinetic models while preserving equivalency between the kinetic models. Using this workflow, we demonstrate that model complexity in terms of networks size has significant impact on sensitivity characteristics of kinetic models. Therefore, it is essential to account for the effects of network complexity when constructing kinetic models. The presented metric for measuring MSC sensitivity to structural changes can guide modelers and experimentalists in improving model quality and guide synthetic biology and metabolic engineering. Our proposed workflow enables the testing of the suitability of a kinetic model for answering certain study-specific questions. We argue that the model-based metabolic design targets that are common across models of different size are of higher confidence, while those that are different could be the objective of investigations for model improvement.
在过去的二十年中,人们在构建细胞代谢的大规模动力学模型方面做出了巨大努力。然而,迄今为止发表的大多数动力学模型仍然集中在中心碳途径周围,或者是围绕特定的简化模型构建的,而没有明确说明其推导和使用的依据。存在用于将基因组规模的代谢重建简化为构建热力学可行且一致简化的代谢模型的系统算法。然而,在我们将它们应用于研究生物系统之前,有必要研究网络复杂性如何影响基于一致简化模型构建的大规模动力学模型得出的结论。
我们系统地简化了 iJO1366 大肠杆菌基因组规模的代谢重建,以构建三个不同大小的代谢模型。由于简化模型是围绕进行简化的核心子系统展开的扩展,因此这些模型是嵌套的。我们提出了一种从最小模型扩展通量分布和浓度向量参考稳定态到较大模型的方法,同时保持最大等效性。围绕参考稳定态和代谢敏感性系数(MSC)构建了动力学模型种群,并计算了它们的代谢敏感性系数。MSC 对模型复杂性敏感。我们提出了一种用于测量 MSC 对这些结构变化的敏感性的度量。
我们首次提出了一种在保持动力学模型等效性的同时扩展模型大小的工作流程。使用此工作流程,我们证明了网络大小的模型复杂性对动力学模型的敏感性特征有重大影响。因此,在构建动力学模型时,必须考虑网络复杂性的影响。用于测量 MSC 对结构变化的敏感性的度量可以指导建模者和实验者提高模型质量,并指导合成生物学和代谢工程。我们提出的工作流程可以测试动力学模型是否适合回答特定研究问题。我们认为,不同大小模型共有的基于模型的代谢设计目标更可信,而那些不同的目标可能是模型改进的调查目标。