Haghbakhsh Reza, Raeissi Sona, Duarte Ana Rita C
LAQV, REQUIMTE, Departamento de Química da Faculdade de Ciências e Tecnologia, Universidade Nova de Lisboa, 2829-516, Caparica, Portugal.
School of Chemical and Petroleum Engineering, Shiraz University, Mollasadra Ave., 71348-51154, Shiraz, Iran.
Sci Rep. 2021 Mar 23;11(1):6684. doi: 10.1038/s41598-021-85824-z.
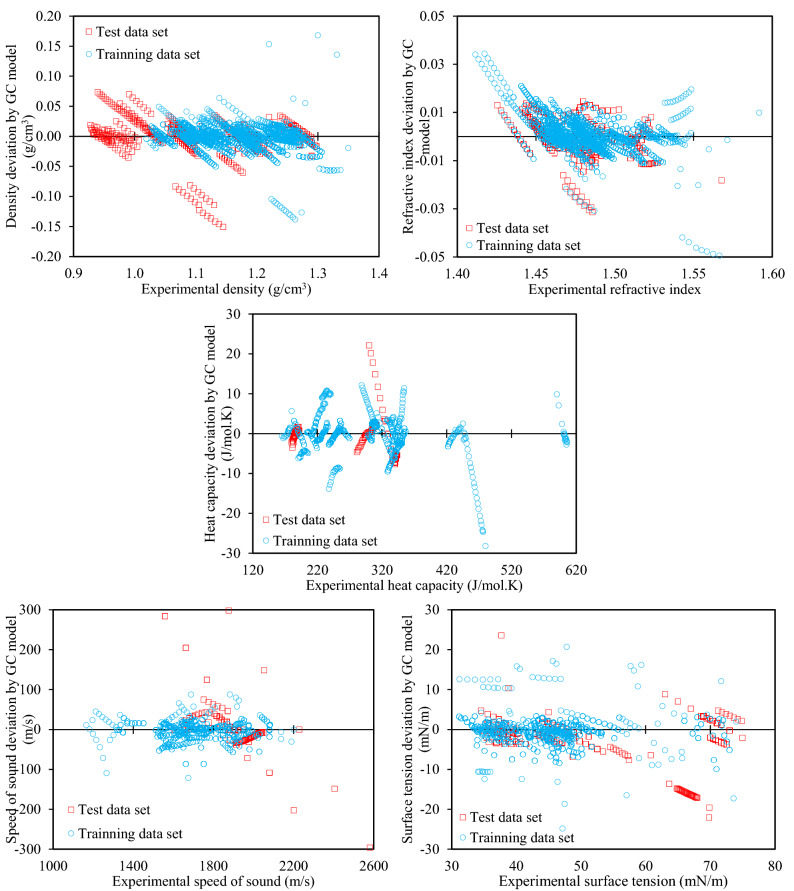
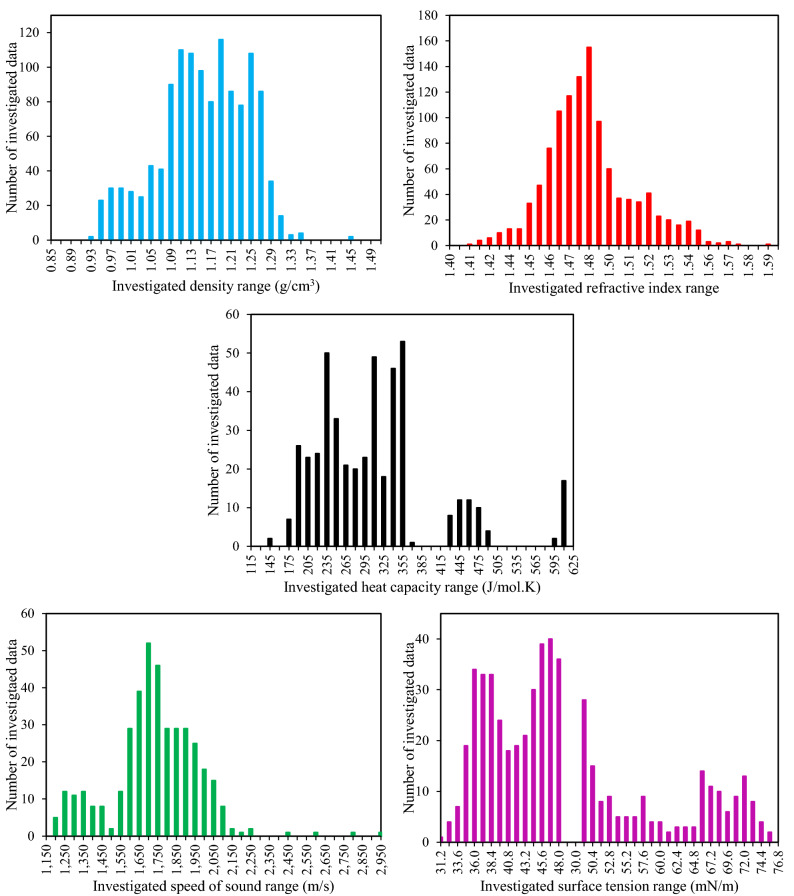
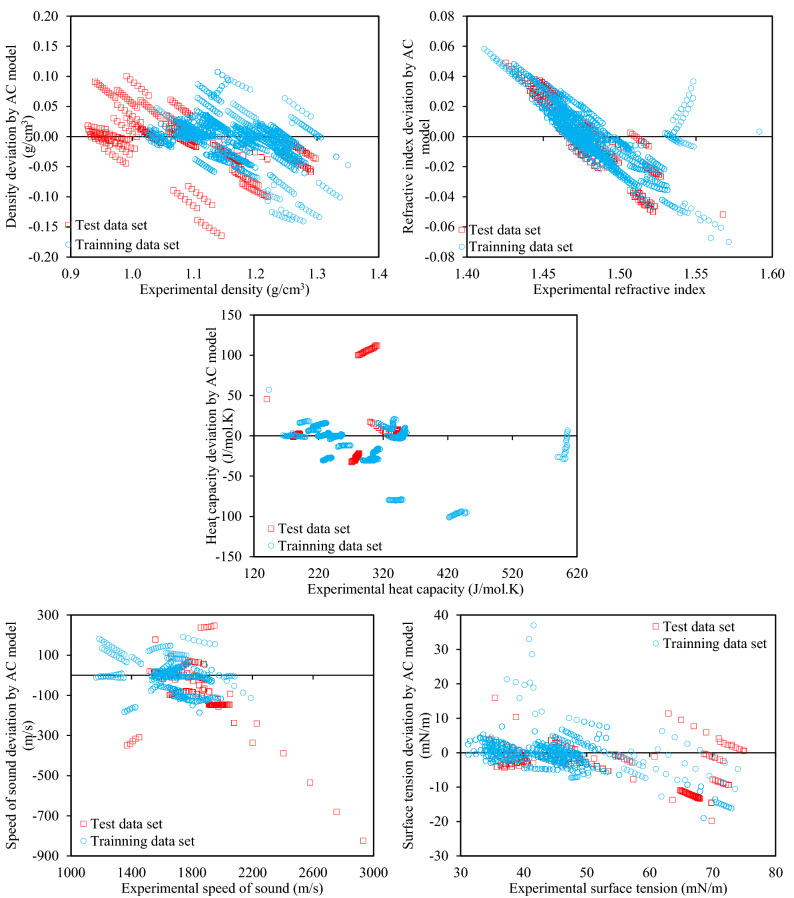
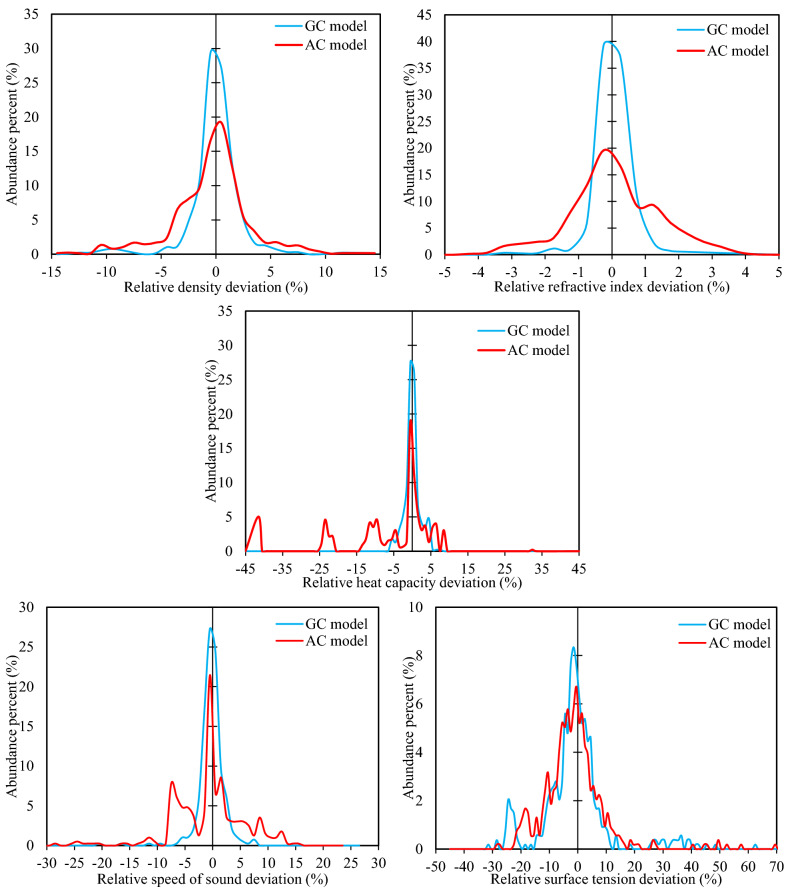
The urgency of advancing green chemistry from labs and computers into the industries is well-known. The Deep Eutectic Solvents (DESs) are a promising category of novel green solvents which simultaneously have the best advantages of liquids and solids. Furthermore, they can be designed or engineered to have the characteristics desired for a given application. However, since they are rather new, there are no general models available to predict the properties of DESs without requiring other properties as input. This is particularly a setback when screening is required for feasibility studies, since a vast number of DESs are envisioned. For the first time, this study presents five group contribution (GC) and five atomic contribution (AC) models for densities, refractive indices, heat capacities, speeds of sound, and surface tensions of DESs. The models, developed using the most up-to-date databank of various types of DESs, simply decompose the molecular structure into a number of predefined groups or atoms. The resulting AARD% of densities, refractive indices, heat capacities, speeds of sound and surface tensions were, respectively, 1.44, 0.37, 3.26, 1.62, and 7.59% for the GC models, and 2.49, 1.03, 9.93, 4.52 and 7.80% for the AC models. Perhaps, even more importantly for designer solvents, is the predictive capability of the models, which was also shown to be highly reliable. Accordingly, very simple, yet highly accurate models are provided that are global for DESs and needless of any physical property information, making them useful predictive tools for a category of green solvents, which is only starting to show its potentials in green technology.
将绿色化学从实验室和计算机推进到工业领域的紧迫性是众所周知的。深共晶溶剂(DESs)是一类很有前景的新型绿色溶剂,它同时具备液体和固体的最佳优点。此外,它们可以被设计或改造以具备特定应用所需的特性。然而,由于它们相当新,目前还没有通用模型可用于在不需要其他性质作为输入的情况下预测DESs的性质。当可行性研究需要进行筛选时,这尤其不利,因为设想了大量的DESs。本研究首次提出了五个基团贡献(GC)模型和五个原子贡献(AC)模型,用于预测DESs的密度、折射率、热容、声速和表面张力。这些模型是利用各类DESs的最新数据库开发的,只需将分子结构分解为一些预定义的基团或原子。GC模型得到的密度、折射率、热容、声速和表面张力的平均绝对相对偏差(AARD%)分别为1.44%、0.37%、3.26%、1.62%和7.59%,AC模型的分别为2.49%、1.03%、9.93%、4.52%和7.80%。也许,对于定制溶剂来说更重要的是这些模型的预测能力,事实也证明其高度可靠。因此,提供了非常简单但高度准确的全局适用于DESs的模型,且无需任何物理性质信息,使其成为一类绿色溶剂的有用预测工具,这类绿色溶剂才刚刚开始在绿色技术中展现其潜力。