Center for Genome Research, University of Modena and Reggio Emilia, 41125 Modena, Italy.
Department of Medical and Surgical Sciences, University of Modena and Reggio Emilia, 41124 Modena, Italy.
Genes (Basel). 2021 Mar 8;12(3):384. doi: 10.3390/genes12030384.
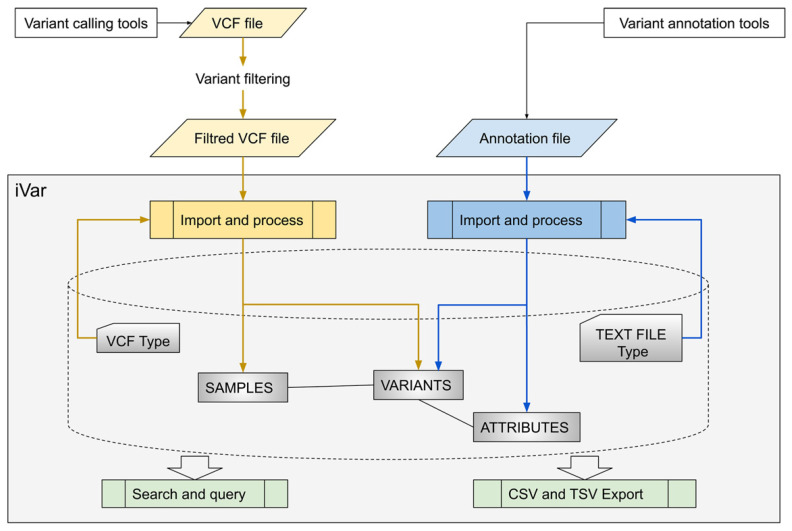
The rapid evolution of Next Generation Sequencing in clinical settings, and the resulting challenge of variant reinterpretation given the constantly updated information, require robust data management systems and organized approaches. In this paper, we present iVar: a freely available and highly customizable tool with a user-friendly web interface. It represents a platform for the unified management of variants identified by different sequencing technologies. iVar accepts variant call format (VCF) files and text annotation files and elaborates them, optimizing data organization and avoiding redundancies. Updated annotations can be periodically re-uploaded and associated with variants as historically tracked attributes, i.e., modifications can be recorded whenever an updated value is imported, thus keeping track of all changes. Data can be visualized through variant-centered and sample-centered interfaces. A customizable search function can be exploited to periodically check if pathogenicity-related data of a variant has changed over time. Patient recontacting ensuing from variant reinterpretation is made easier by iVar through the effective identification of all patients present in the database carrying a specific variant. We tested iVar by uploading 4171 VCF files and 1463 annotation files, obtaining a database of 4166 samples and 22,569 unique variants. iVar has proven to be a useful tool with good performance in terms of collecting and managing data from a medium-throughput laboratory.
下一代测序技术在临床环境中的快速发展,以及不断更新的信息导致的变异重新解释的挑战,都需要强大的数据管理系统和有组织的方法。在本文中,我们介绍了 iVar:这是一个免费且高度可定制的工具,具有用户友好的网络界面。它是一个用于统一管理不同测序技术识别出的变异的平台。iVar 接受变异调用格式 (VCF) 文件和文本注释文件,并对其进行详细分析,优化数据组织并避免冗余。定期重新上传更新的注释,并将其作为历史跟踪属性与变体相关联,即每当导入更新的值时,可以记录修改,从而跟踪所有更改。可以通过变体中心和样本中心界面查看数据。可以利用可定制的搜索功能定期检查变体的致病性相关数据是否随时间发生变化。通过 iVar,通过有效识别数据库中携带特定变异的所有患者,更容易实现对变异重新解释的患者重新联系。我们通过上传 4171 个 VCF 文件和 1463 个注释文件对 iVar 进行了测试,获得了一个包含 4166 个样本和 22569 个独特变体的数据库。iVar 已被证明是一种有用的工具,在从中等通量实验室收集和管理数据方面表现良好。