Department of Oncology, Ludwig Institute for Cancer Research Lausanne, Lausanne University Hospital and the University of Lausanne, Lausanne, Switzerland.
SIB Swiss Institute of Bioinformatics, Lausanne, Switzerland.
Mol Cell Proteomics. 2021;20:100080. doi: 10.1016/j.mcpro.2021.100080. Epub 2021 Apr 9.
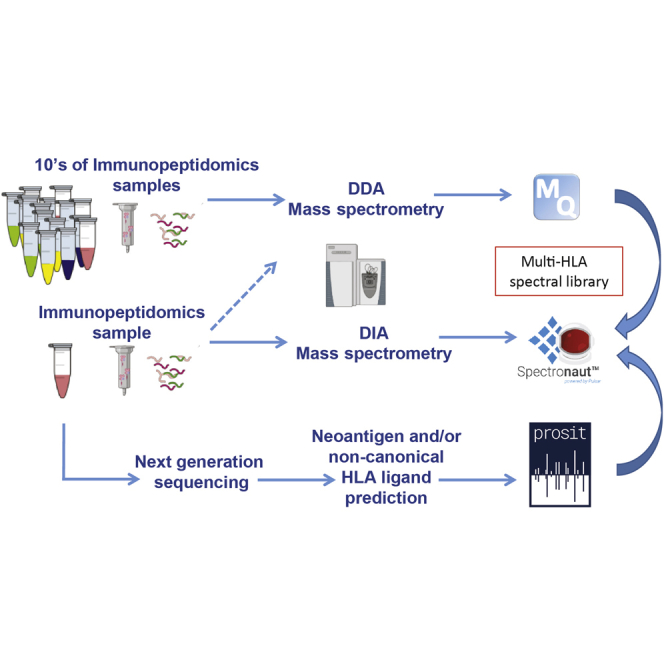
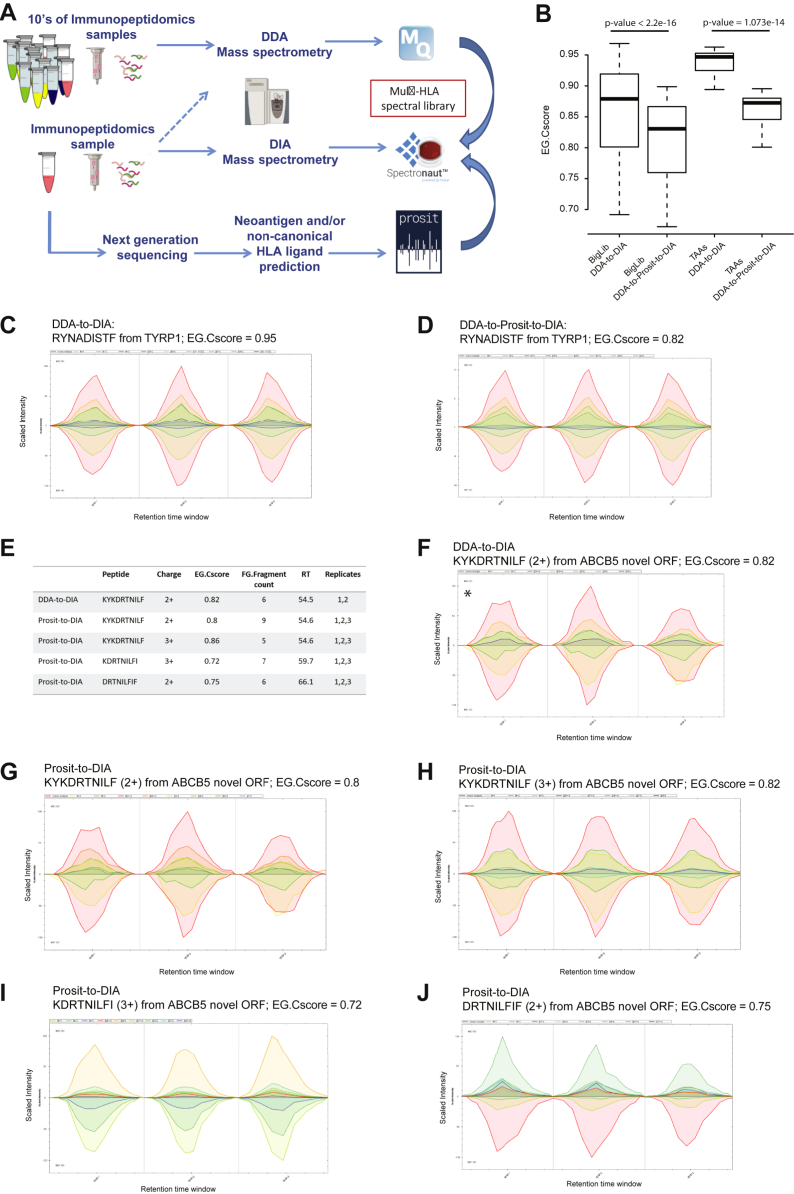
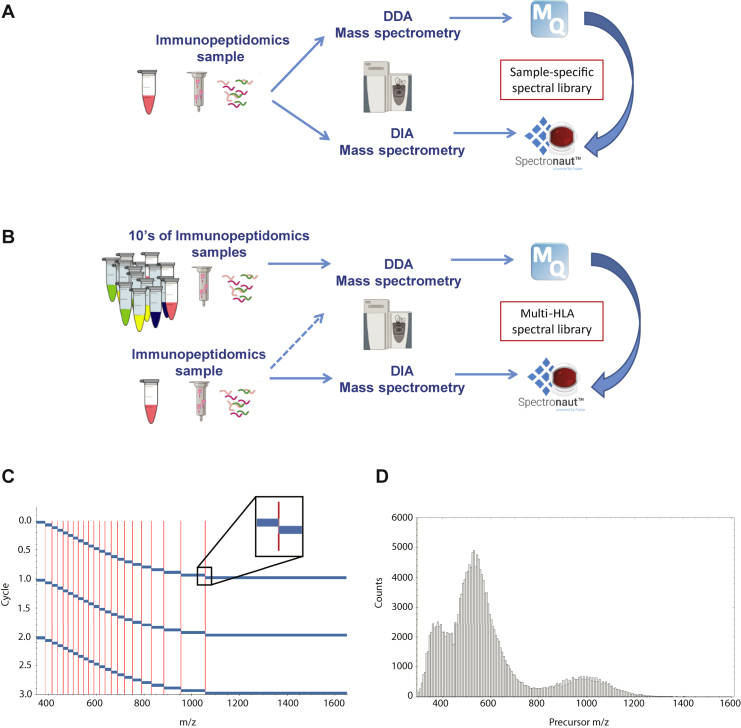
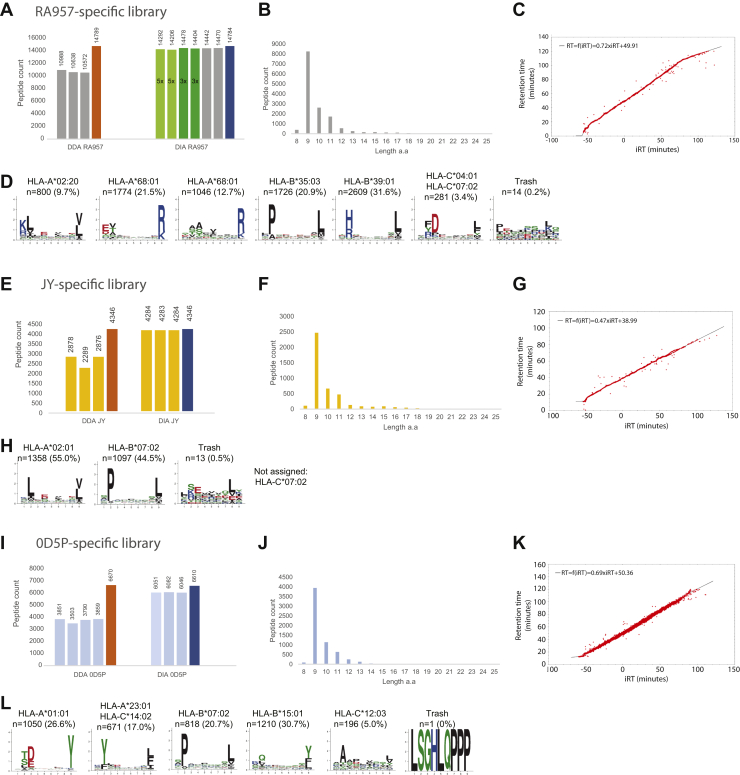
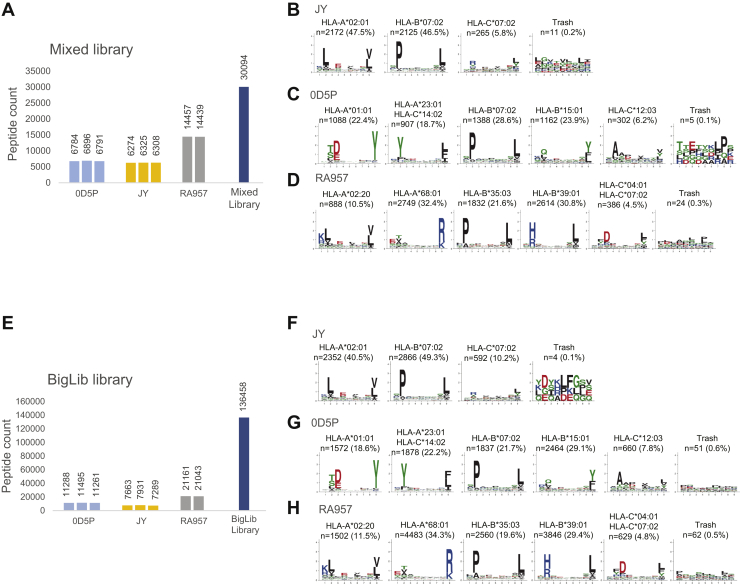
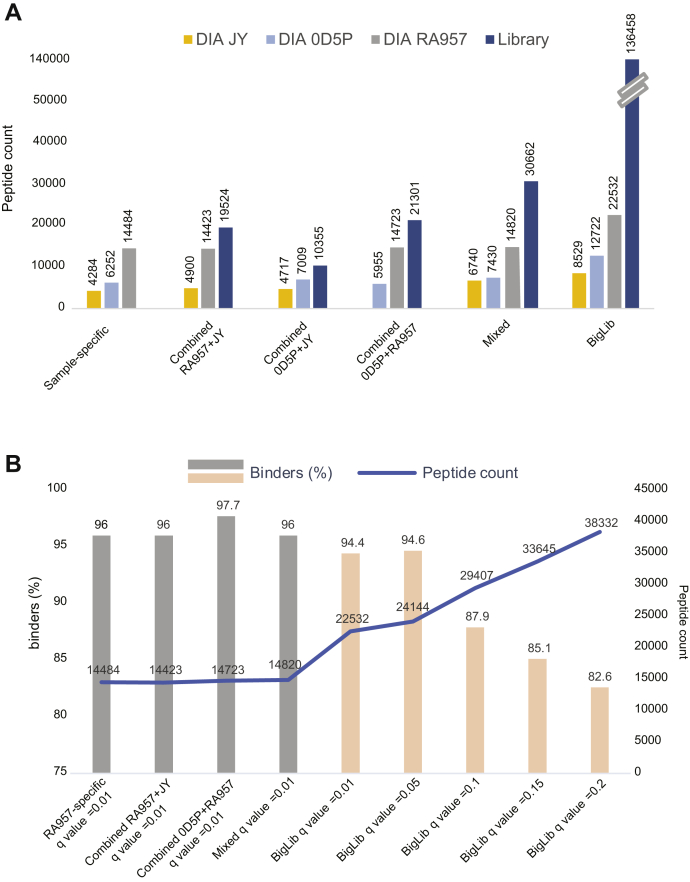
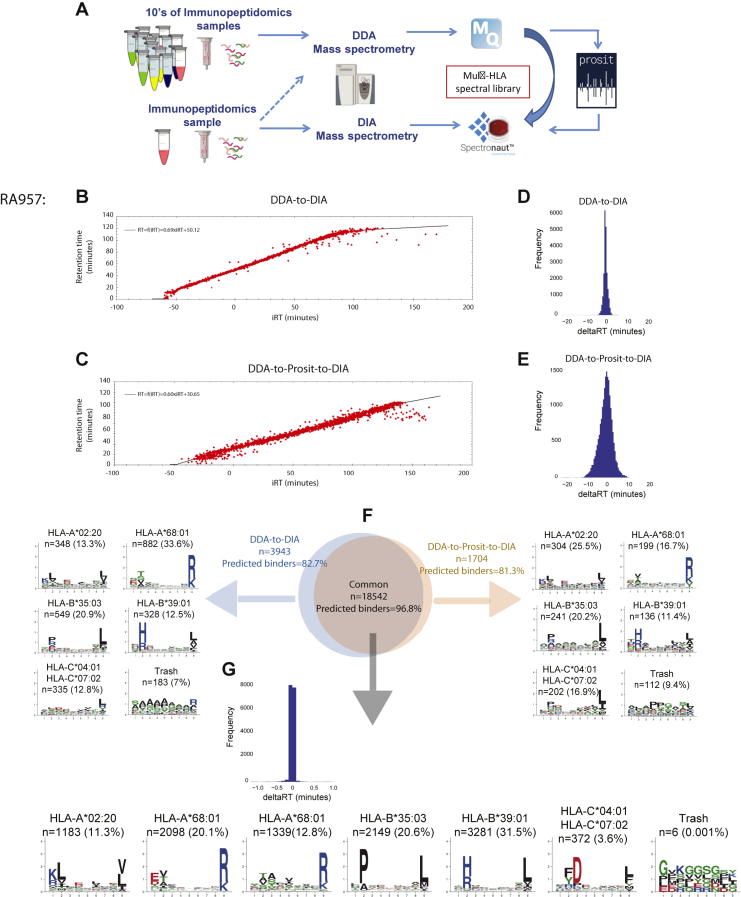
Mass spectrometry (MS) is the state-of-the-art methodology for capturing the breadth and depth of the immunopeptidome across human leukocyte antigen (HLA) allotypes and cell types. The majority of studies in the immunopeptidomics field are discovery driven. Hence, data-dependent tandem MS (MS/MS) acquisition (DDA) is widely used, as it generates high-quality references of peptide fingerprints. However, DDA suffers from the stochastic selection of abundant ions that impairs sensitivity and reproducibility. In contrast, in data-independent acquisition (DIA), the systematic fragmentation and acquisition of all fragment ions within given isolation m/z windows yield a comprehensive map for a given sample. However, many DIA approaches commonly require generating comprehensive DDA-based spectrum libraries, which can become impractical for studying noncanonical and personalized neoantigens. Because the amount of HLA peptides eluted from biological samples such as small tissue biopsies is typically not sufficient for acquiring both meaningful DDA data necessary for generating comprehensive spectral libraries and DIA MS measurements, the implementation of DIA in the immunopeptidomics translational research domain has remained limited. We implemented a DIA immunopeptidomics workflow and assessed its sensitivity and accuracy by matching DIA data against libraries with growing complexity-from sample-specific libraries to libraries combining 2 to 40 different immunopeptidomics samples. Analyzing DIA immunopeptidomics data against a complex multi-HLA spectral library resulted in a two-fold increase in peptide identification compared with sample-specific library and in a three-fold increase compared with DDA measurements, yet with no detrimental effect on the specificity. Furthermore, we demonstrated the implementation of DIA for sensitive personalized neoantigen discovery through the analysis of DIA data with predicted MS/MS spectra of clinically relevant HLA ligands. We conclude that a comprehensive multi-HLA library for DIA approach in combination with MS/MS prediction is highly advantageous for clinical immunopeptidomics, especially when low amounts of biological samples are available.
质谱(MS)是捕获人类白细胞抗原(HLA)同种型和细胞类型的免疫肽组广度和深度的最先进方法。免疫肽组学领域的大多数研究都是以发现为驱动的。因此,广泛使用依赖于数据的串联 MS(MS/MS)采集(DDA),因为它生成了高质量的肽指纹参考。然而,DDA 受到大量离子随机选择的影响,从而损害了其灵敏度和重现性。相比之下,在独立于数据的采集(DIA)中,系统地碎片化和获取给定隔离 m/z 窗口内的所有碎片离子,为给定样本生成全面的图谱。然而,许多 DIA 方法通常需要生成全面的基于 DDA 的谱库,对于研究非规范和个性化的新抗原,这可能变得不切实际。因为从小组织活检等生物样本中洗脱的 HLA 肽的量通常不足以同时获取生成全面谱库和 DIA MS 测量所需的有意义的 DDA 数据,因此 DIA 在免疫肽组学转化研究领域的实施仍然受到限制。我们实施了 DIA 免疫肽组学工作流程,并通过将 DIA 数据与具有不断增加复杂性的库进行匹配来评估其灵敏度和准确性,从特定于样本的库到结合 2 到 40 个不同免疫肽组学样本的库。与复杂的多 HLA 光谱库相比,分析 DIA 免疫肽组学数据可使肽鉴定增加两倍,与特定于样本的库相比增加三倍,而对特异性没有不利影响。此外,我们通过分析具有临床相关 HLA 配体的预测 MS/MS 光谱的 DIA 数据,展示了 DIA 在敏感的个性化新抗原发现中的实施。我们得出结论,DIA 方法的全面多 HLA 库与 MS/MS 预测相结合,对于临床免疫肽组学非常有利,特别是当可用的生物样本量较少时。