School of Mathematical Sciences and LPMC, Nankai University, Tianjin, 300071, China.
MOE Key Laboratory of Bioinformatics and Bioinformatics Division of BNRIST, Department of Automation, Tsinghua University, 100084, Beijing, China.
Nat Commun. 2024 Feb 22;15(1):1629. doi: 10.1038/s41467-024-46045-w.
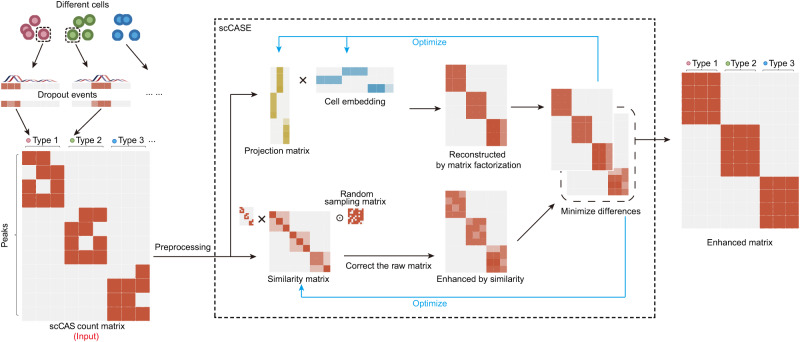
Single-cell chromatin accessibility sequencing (scCAS) has emerged as a valuable tool for interrogating and elucidating epigenomic heterogeneity and gene regulation. However, scCAS data inherently suffers from limitations such as high sparsity and dimensionality, which pose significant challenges for downstream analyses. Although several methods are proposed to enhance scCAS data, there are still challenges and limitations that hinder the effectiveness of these methods. Here, we propose scCASE, a scCAS data enhancement method based on non-negative matrix factorization which incorporates an iteratively updating cell-to-cell similarity matrix. Through comprehensive experiments on multiple datasets, we demonstrate the advantages of scCASE over existing methods for scCAS data enhancement. The interpretable cell type-specific peaks identified by scCASE can provide valuable biological insights into cell subpopulations. Moreover, to leverage the large compendia of available omics data as a reference, we further expand scCASE to scCASER, which enables the incorporation of external reference data to improve enhancement performance.
单细胞染色质可及性测序 (scCAS) 已成为探究和阐明表观基因组异质性和基因调控的有价值的工具。然而,scCAS 数据本质上存在着高稀疏性和高维度等限制,这给下游分析带来了重大挑战。尽管已经提出了几种方法来增强 scCAS 数据,但仍然存在一些挑战和限制,这些限制阻碍了这些方法的有效性。在这里,我们提出了 scCASE,这是一种基于非负矩阵分解的 scCAS 数据增强方法,它结合了一个迭代更新的细胞间相似性矩阵。通过对多个数据集的综合实验,我们证明了 scCASE 在增强 scCAS 数据方面优于现有的方法。scCASE 识别的可解释的细胞类型特异性峰可以为细胞亚群提供有价值的生物学见解。此外,为了利用大量现有的组学数据作为参考,我们进一步扩展了 scCASE 到 scCASER,它能够整合外部参考数据来提高增强性能。