Department of Chemistry, NC State University, Raleigh, NC, USA.
Comparative Medicine Institute, NC State University, Raleigh, NC, USA.
Nat Commun. 2021 Apr 13;12(1):2193. doi: 10.1038/s41467-021-22497-2.
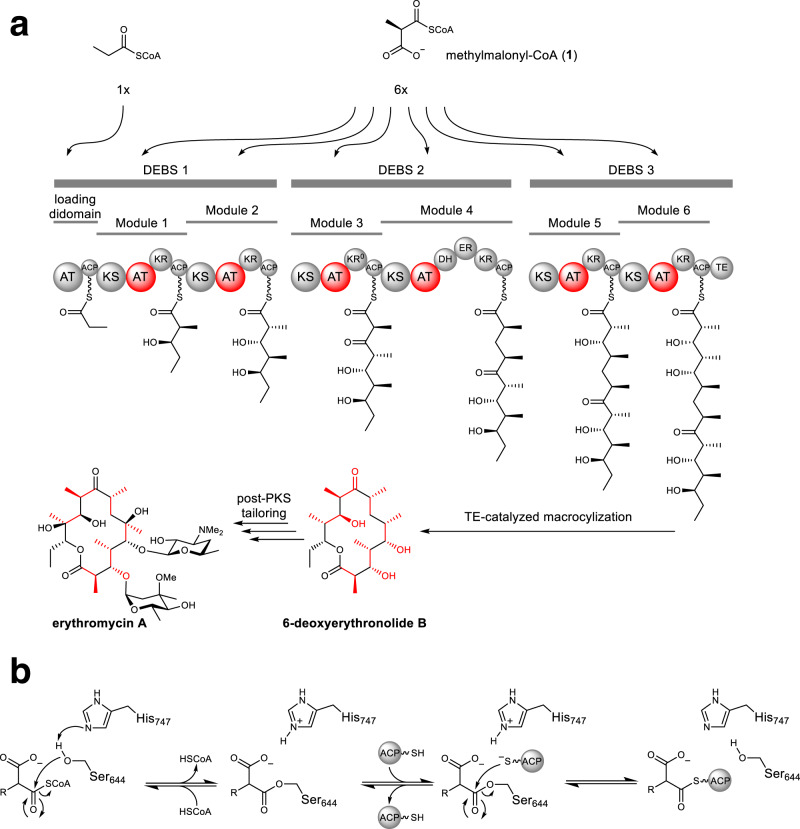
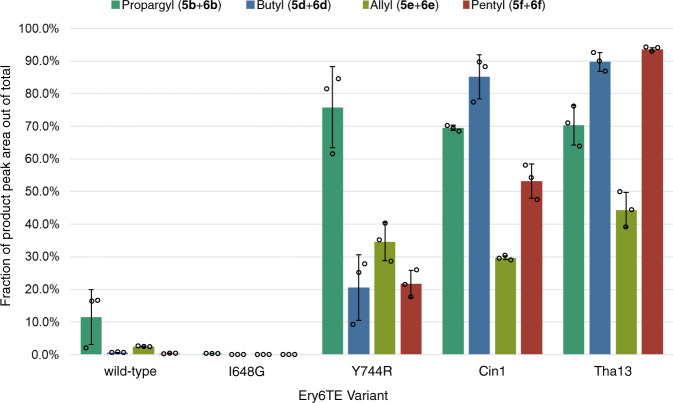
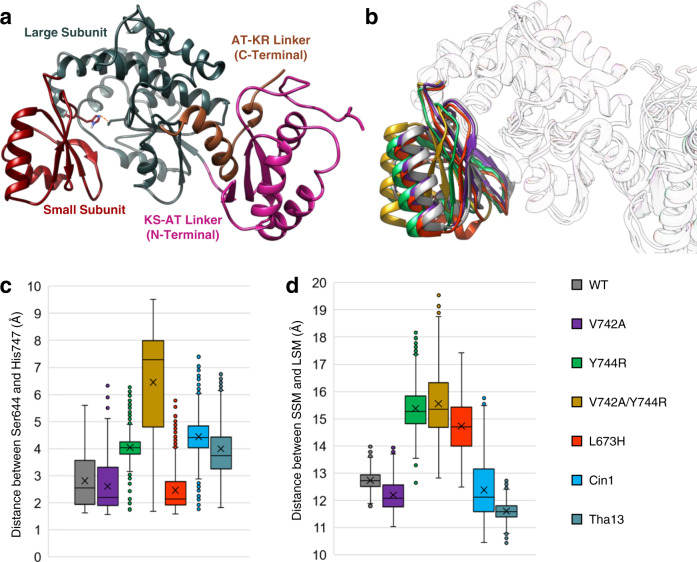
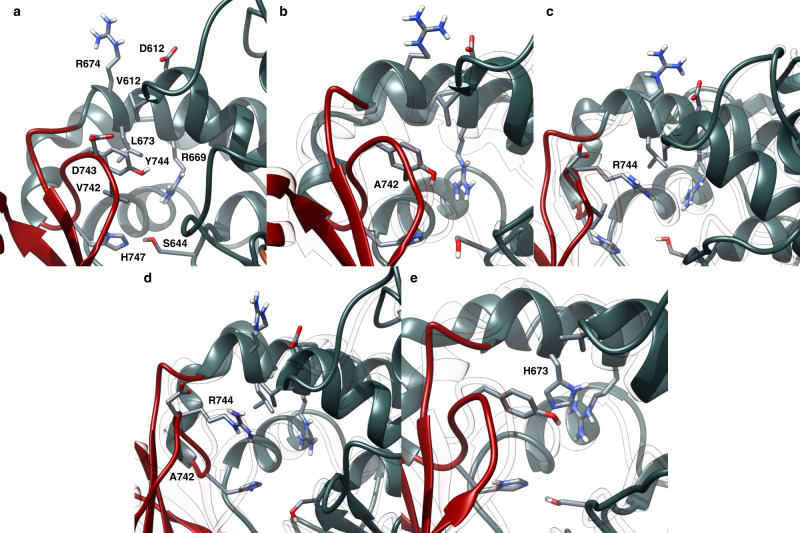
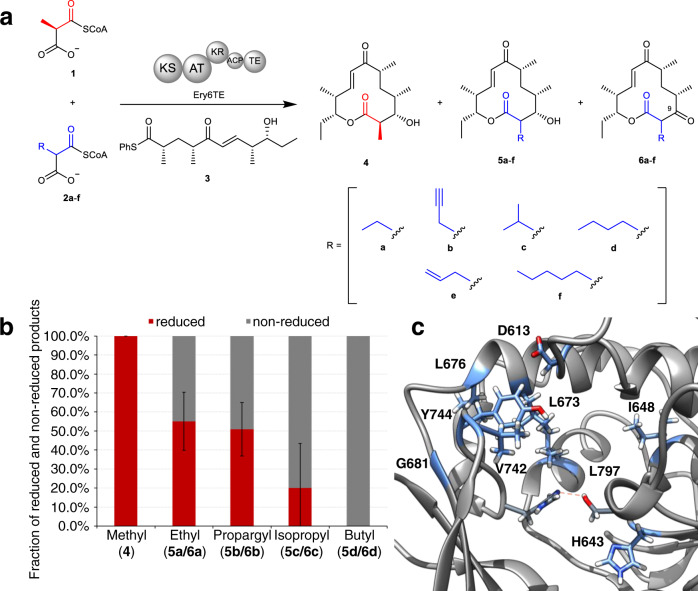
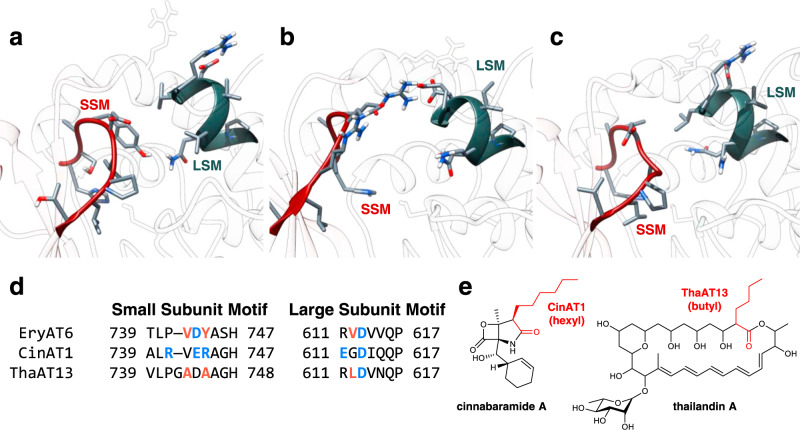
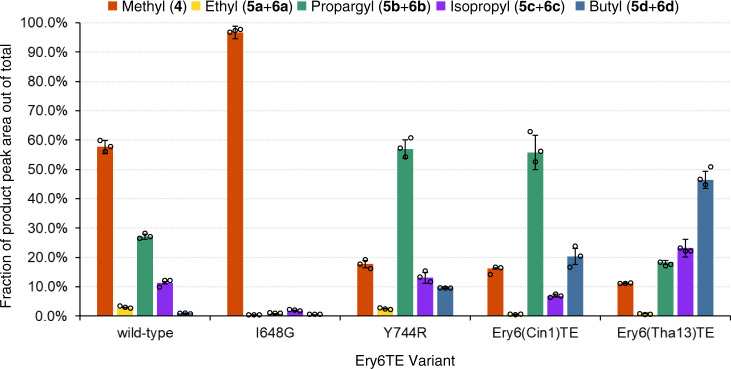
Polyketides, one of the largest classes of natural products, are often clinically relevant. The ability to engineer polyketide biosynthesis to produce analogs is critically important. Acyltransferases (ATs) of modular polyketide synthases (PKSs) catalyze the installation of malonyl-CoA extenders into polyketide scaffolds. ATs have been targeted extensively to site-selectively introduce various extenders into polyketides. Yet, a complete inventory of AT residues responsible for substrate selection has not been established, limiting the scope of AT engineering. Here, molecular dynamics simulations are used to prioritize ~50 mutations within the active site of EryAT6 from erythromycin biosynthesis, leading to identification of two previously unexplored structural motifs. Exchanging both motifs with those from ATs with alternative extender specificities provides chimeric PKS modules with expanded and inverted substrate specificity. Our enhanced understanding of AT substrate selectivity and application of this motif-swapping strategy are expected to advance our ability to engineer PKSs towards designer polyketides.
聚酮类化合物是最大的天然产物类别之一,通常具有临床相关性。能够对聚酮生物合成进行工程改造以生产类似物是至关重要的。多功能聚酮合酶(PKS)中的酰基转移酶(AT)催化将丙二酰辅酶 A 延长物装入聚酮骨架中。AT 已被广泛靶向以选择性地将各种延长物引入聚酮中。然而,尚未建立负责底物选择的完整 AT 残基清单,限制了 AT 工程的范围。在这里,使用分子动力学模拟对红霉素生物合成中 EryAT6 的活性位点中的约 50 个突变进行优先级排序,从而鉴定出两个以前未探索过的结构基序。将这两个基序与具有替代延长特异性的 AT 的基序进行交换,为具有扩展和反转底物特异性的嵌合 PKS 模块提供了可能性。我们对 AT 底物选择性的增强理解和应用这种基序交换策略有望提高我们对 PKS 进行工程改造以获得设计性聚酮的能力。