Lester and Sue Smith Breast Center, Baylor College of Medicine, Houston, TX, USA; Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX, USA.
Department of Liver Surgery and Transplantation, Liver Cancer Institute, Zhongshan Hospital, Fudan University, and Key Laboratory of Carcinogenesis and Cancer Invasion of Ministry of Education, Shanghai, China.
Mol Cell Proteomics. 2021;20:100083. doi: 10.1016/j.mcpro.2021.100083. Epub 2021 Apr 20.
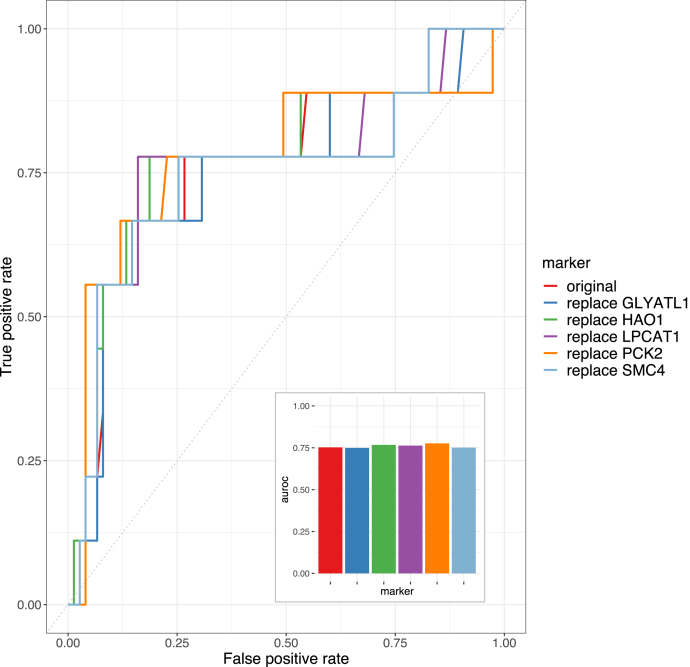
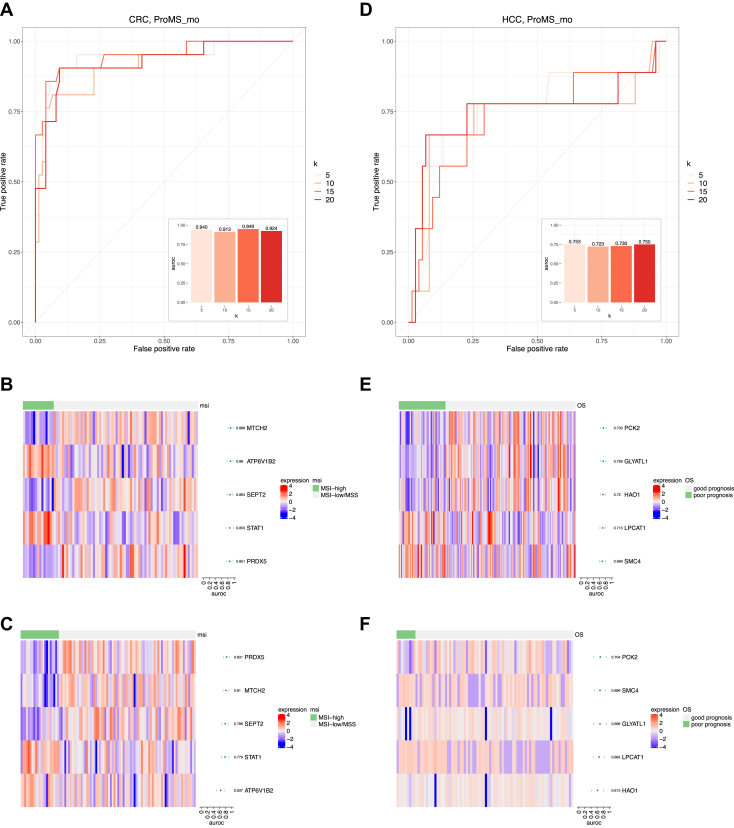
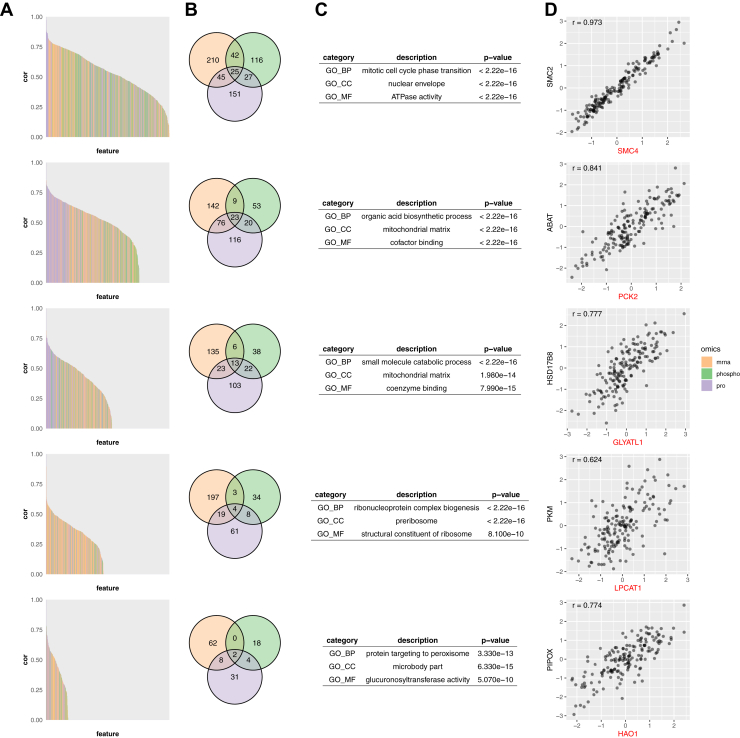
Untargeted mass spectrometry (MS)-based proteomics provides a powerful platform for protein biomarker discovery, but clinical translation depends on the selection of a small number of proteins for downstream verification and validation. Due to the small sample size of typical discovery studies, protein markers identified from discovery data may not be generalizable to independent datasets. In addition, a good protein marker identified using a discovery platform may be difficult to implement in verification and validation platforms. Moreover, although multiomics characterization is being increasingly used in discovery cohort studies, there is no existing method for multiomics-facilitated protein biomarker selection. Here, we present ProMS, a computational algorithm for protein marker selection. The algorithm is based on the hypothesis that a phenotype is characterized by a few underlying biological functions, each manifested by a group of coexpressed proteins. A weighted k-medoids clustering algorithm is applied to all univariately informative proteins to identify both coexpressed protein clusters and a representative protein for each cluster as markers. In two clinically important classification problems, ProMS shows superior performance compared with existing feature selection methods. ProMS can be extended to the multiomics setting (ProMS_mo) through a constrained weighted k-medoids clustering algorithm, and the protein panels selected by ProMS_mo show improved performance on independent test data compared with ProMS. In addition to superior performance, ProMS and ProMS_mo also have two unique strengths. First, the feature clusters enable functional interpretation of the selected protein markers. Second, the feature clusters provide an opportunity to select replacement protein markers, facilitating a robust transition to the verification and validation platforms. In summary, this study provides a unified and effective computational framework for selecting protein biomarkers using proteomics or multiomics data. The software implementation is publicly available at https://github.com/bzhanglab/proms.
非靶向质谱(MS)- 基于蛋白质组学为蛋白质生物标志物的发现提供了一个强大的平台,但临床转化取决于对少量蛋白质进行下游验证和确认的选择。由于典型发现研究的样本量较小,因此从发现数据中识别的蛋白质标志物可能无法推广到独立数据集。此外,使用发现平台识别的良好蛋白质标志物可能难以在验证和确认平台中实施。此外,尽管多组学特征在发现队列研究中越来越多地使用,但目前尚无用于多组学辅助蛋白质生物标志物选择的方法。在这里,我们提出了 ProMS,这是一种用于蛋白质标志物选择的计算算法。该算法基于这样的假设,即表型由少数潜在的生物学功能来特征化,每个功能由一组共表达的蛋白质来表现。应用加权 k 均值聚类算法对所有单变量信息丰富的蛋白质进行分析,以识别共表达蛋白质簇和每个簇的代表性蛋白质作为标志物。在两个具有临床重要性的分类问题中,ProMS 与现有的特征选择方法相比表现出优越的性能。ProMS 可以通过约束加权 k 均值聚类算法扩展到多组学设置(ProMS_mo),并且与 ProMS 相比,ProMS_mo 选择的蛋白质面板在独立测试数据上显示出了更好的性能。除了优越的性能外,ProMS 和 ProMS_mo 还有两个独特的优势。首先,特征聚类使所选蛋白质标志物的功能解释成为可能。其次,特征聚类提供了选择替代蛋白质标志物的机会,从而为可靠地过渡到验证和确认平台提供了机会。总之,本研究为使用蛋白质组学或多组学数据选择蛋白质生物标志物提供了一个统一有效的计算框架。软件实现可在 https://github.com/bzhanglab/proms 上获得。