Department of Systems Biology, Harvard Medical School, Boston, MA, USA.
insitro, South San Francisco, CA, USA.
Nat Commun. 2021 Apr 23;12(1):2403. doi: 10.1038/s41467-021-22732-w.
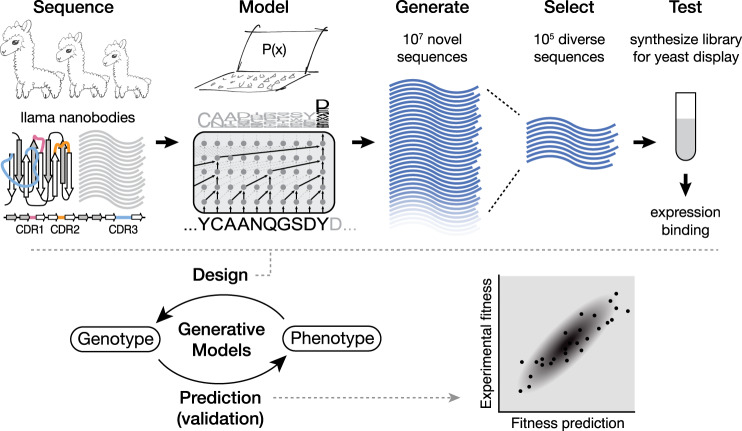
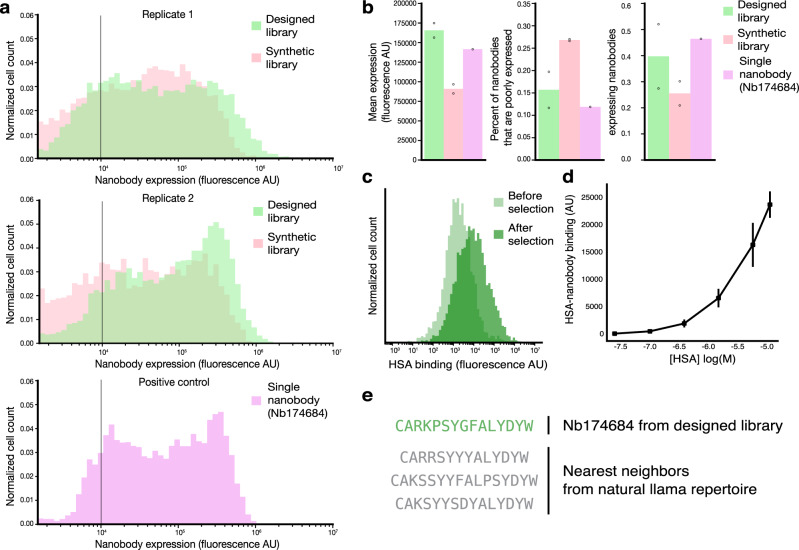
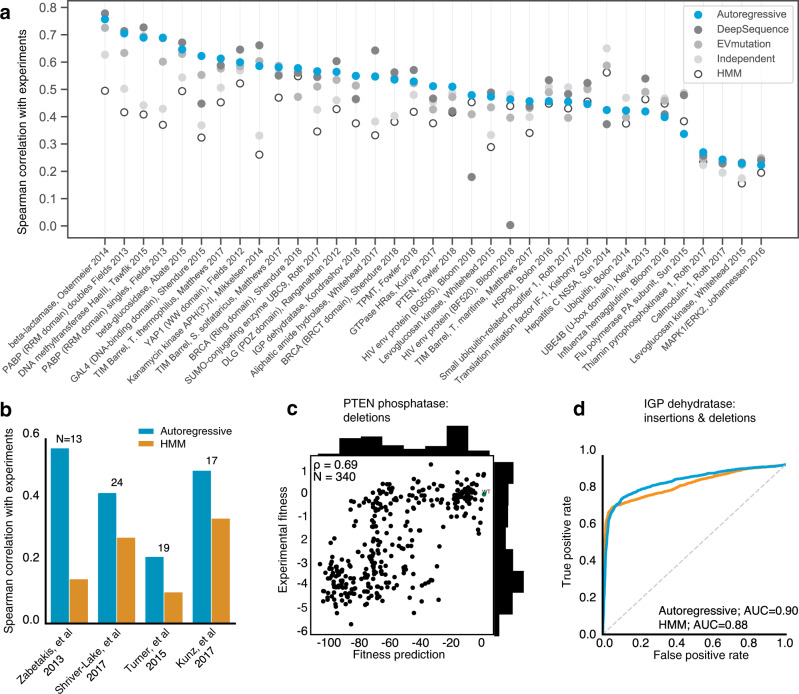
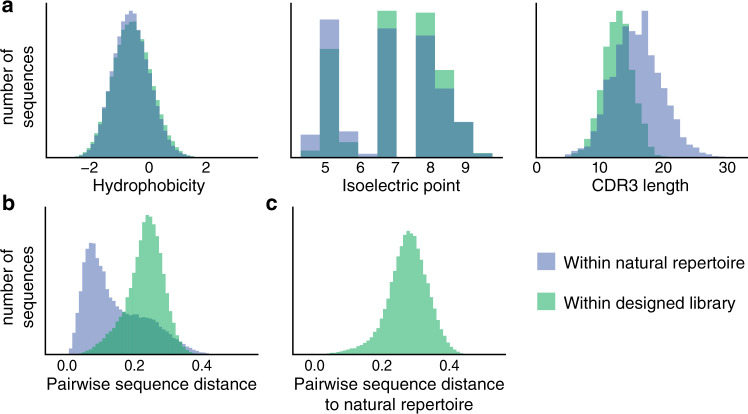
The ability to design functional sequences and predict effects of variation is central to protein engineering and biotherapeutics. State-of-art computational methods rely on models that leverage evolutionary information but are inadequate for important applications where multiple sequence alignments are not robust. Such applications include the prediction of variant effects of indels, disordered proteins, and the design of proteins such as antibodies due to the highly variable complementarity determining regions. We introduce a deep generative model adapted from natural language processing for prediction and design of diverse functional sequences without the need for alignments. The model performs state-of-art prediction of missense and indel effects and we successfully design and test a diverse 10-nanobody library that shows better expression than a 1000-fold larger synthetic library. Our results demonstrate the power of the alignment-free autoregressive model in generalizing to regions of sequence space traditionally considered beyond the reach of prediction and design.
设计功能序列和预测变异影响的能力是蛋白质工程和生物治疗的核心。最先进的计算方法依赖于利用进化信息的模型,但对于某些重要应用来说并不足够,因为这些应用中多序列比对并不稳健。此类应用包括插入缺失、无序蛋白质的变异影响预测,以及由于高度可变的互补决定区而导致的抗体等蛋白质的设计。我们引入了一种源自自然语言处理的深度生成模型,用于在无需比对的情况下预测和设计多样化的功能序列。该模型在预测错义突变和插入缺失影响方面表现出色,我们成功设计并测试了一个多样化的 10 纳米抗体文库,其表达水平优于大 1000 倍的合成文库。我们的结果证明了无比对自回归模型在序列空间的传统认为难以预测和设计的区域进行泛化的能力。