Department of Computer Science, California State University, Los Angeles, CA 90032, USA.
Department of Chemistry and Biochemistry, California State University, Los Angeles, CA 90032, USA.
Molecules. 2021 Apr 20;26(8):2383. doi: 10.3390/molecules26082383.
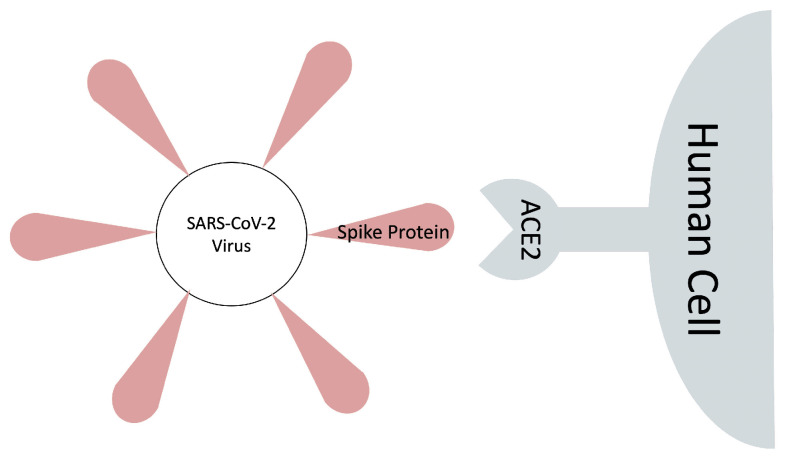
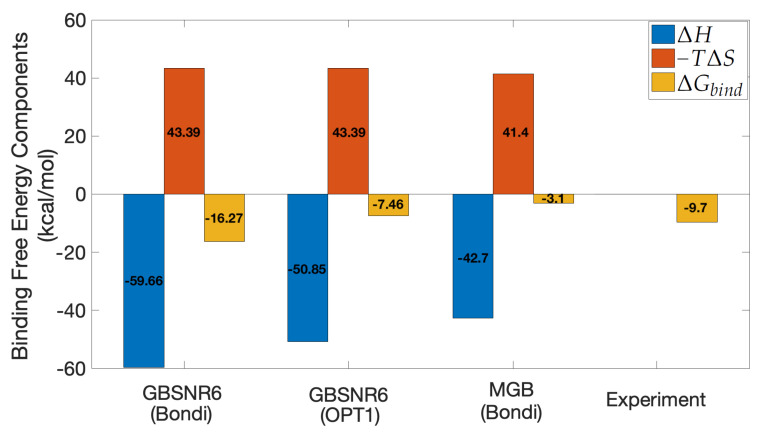
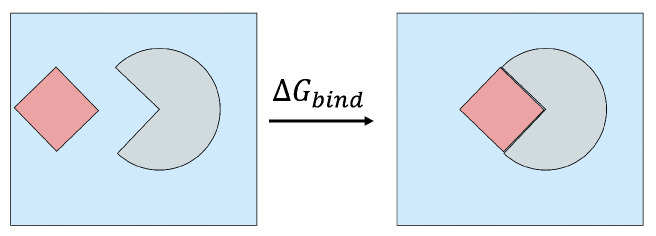
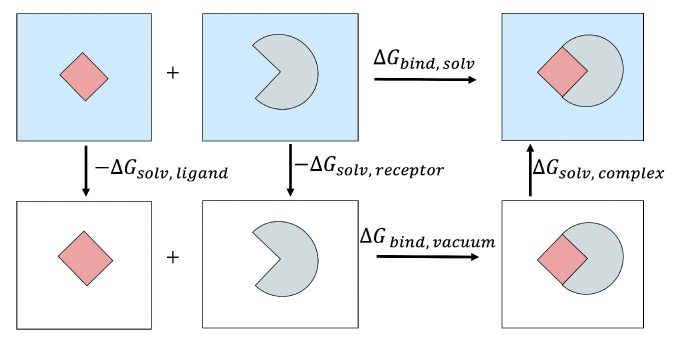
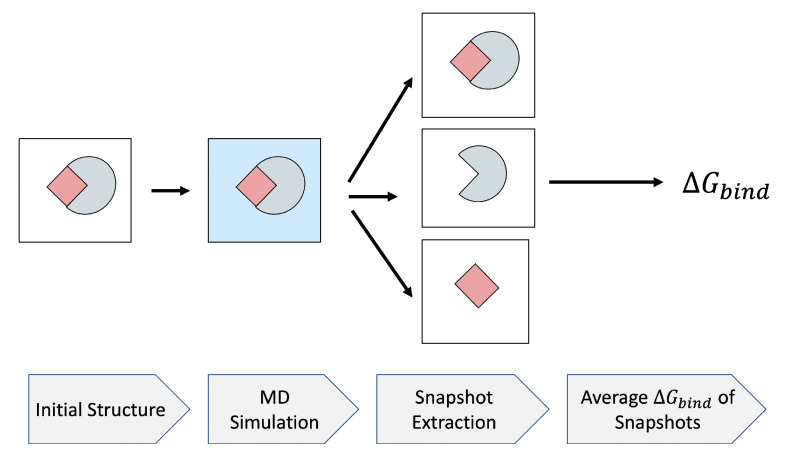
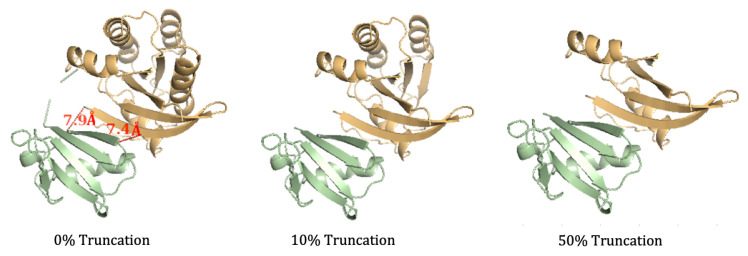
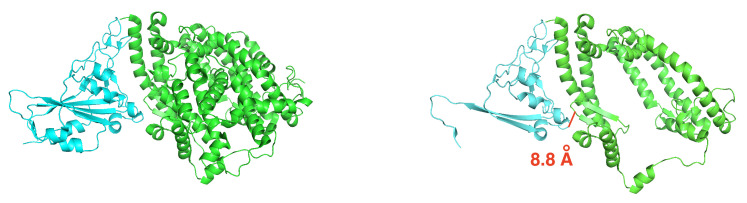
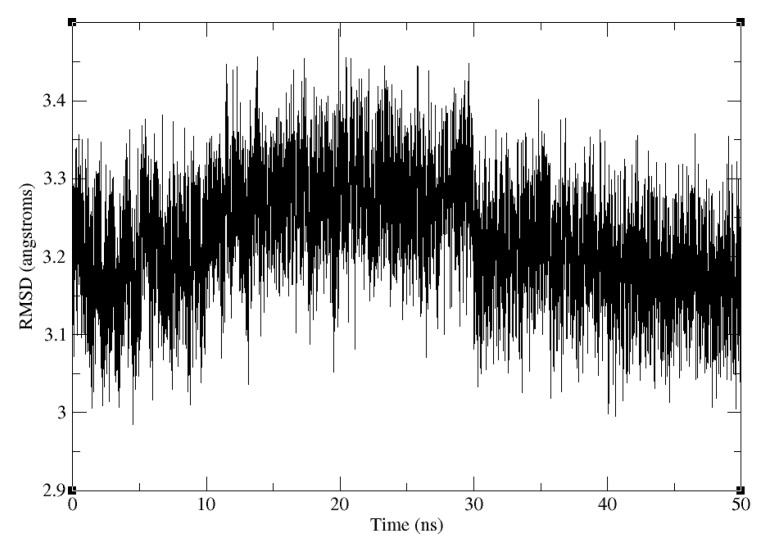
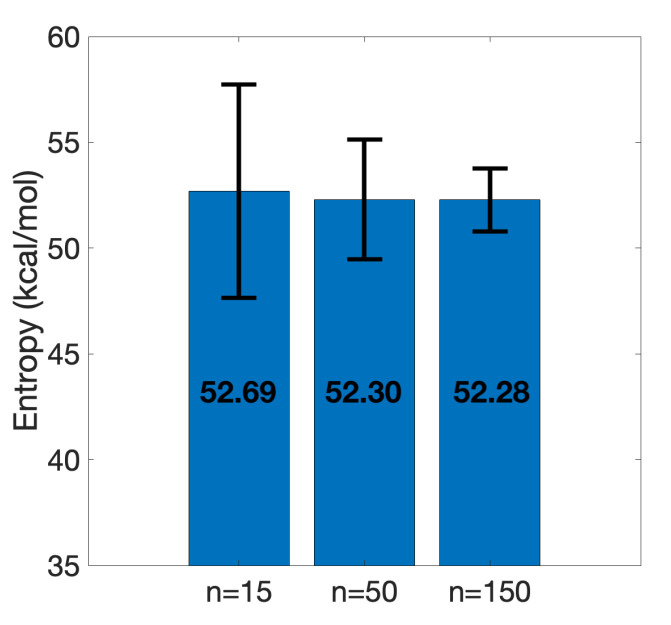
The binding free energy calculation of protein-ligand complexes is necessary for research into virus-host interactions and the relevant applications in drug discovery. However, many current computational methods of such calculations are either inefficient or inaccurate in practice. Utilizing implicit solvent models in the molecular mechanics generalized Born surface area (MM/GBSA) framework allows for efficient calculations without significant loss of accuracy. Here, GBNSR6, a new flavor of the generalized Born model, is employed in the MM/GBSA framework for measuring the binding affinity between SARS-CoV-2 spike protein and the human ACE2 receptor. A computational protocol is developed based on the widely studied Ras-Raf complex, which has similar binding free energy to SARS-CoV-2/ACE2. Two options for representing the dielectric boundary of the complexes are evaluated: one based on the standard Bondi radii and the other based on a newly developed set of atomic radii (OPT1), optimized specifically for protein-ligand binding. Predictions based on the two radii sets provide upper and lower bounds on the experimental references: -14.7(ΔGbindBondi)<-10.6(ΔGbindExp.)<-4.1(ΔGbindOPT1) kcal/mol. The consensus estimates of the two bounds show quantitative agreement with the experiment values. This work also presents a novel truncation method and computational strategies for efficient entropy calculations with normal mode analysis. Interestingly, it is observed that a significant decrease in the number of snapshots does not affect the accuracy of entropy calculation, while it does lower computation time appreciably. The proposed MM/GBSA protocol can be used to study the binding mechanism of new variants of SARS-CoV-2, as well as other relevant structures.
蛋白质-配体复合物的结合自由能计算对于研究病毒-宿主相互作用以及药物发现中的相关应用是必要的。然而,目前许多此类计算的计算方法在实践中要么效率低下,要么不够准确。在分子力学广义 Born 表面面积(MM/GBSA)框架中利用隐溶剂模型可以实现高效计算,而不会显著降低准确性。在这里,我们使用了一种新的广义 Born 模型 GBNSR6,用于测量 SARS-CoV-2 刺突蛋白与人类 ACE2 受体之间的结合亲和力。基于广泛研究的 Ras-Raf 复合物,开发了一种计算方案,该复合物具有与 SARS-CoV-2/ACE2 相似的结合自由能。评估了两种表示复合物介电边界的选项:一种基于标准 Bondi 半径,另一种基于新开发的一组原子半径(OPT1),专门针对蛋白质-配体结合进行了优化。基于这两个半径集的预测为实验参考值提供了上限和下限:-14.7(ΔGbindBondi)<-10.6(ΔGbindExp.)<-4.1(ΔGbindOPT1) kcal/mol。两个边界的共识估计与实验值具有定量一致性。这项工作还提出了一种新的截断方法和计算策略,用于通过正常模式分析进行有效的熵计算。有趣的是,观察到减少快照数量不会影响熵计算的准确性,但会显著降低计算时间。所提出的 MM/GBSA 方案可用于研究 SARS-CoV-2 的新变体以及其他相关结构的结合机制。