Bioinformatics and Computational Biology Program, University of Minnesota, Minneapolis, MN 55455, USA.
Department of Chemistry, Wesleyan University, Middletown, CT 06459, USA.
Int J Mol Sci. 2021 Apr 28;22(9):4619. doi: 10.3390/ijms22094619.
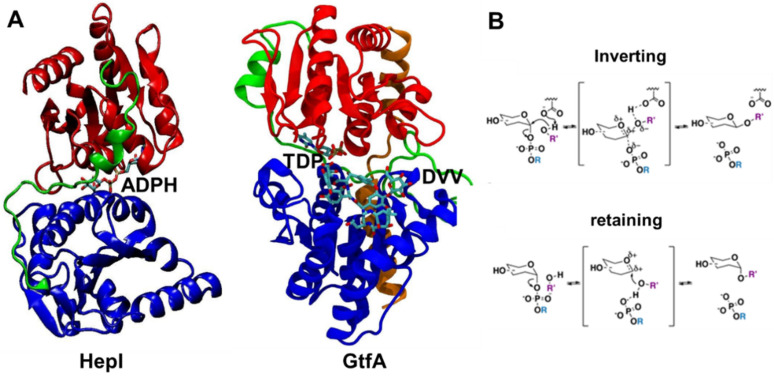
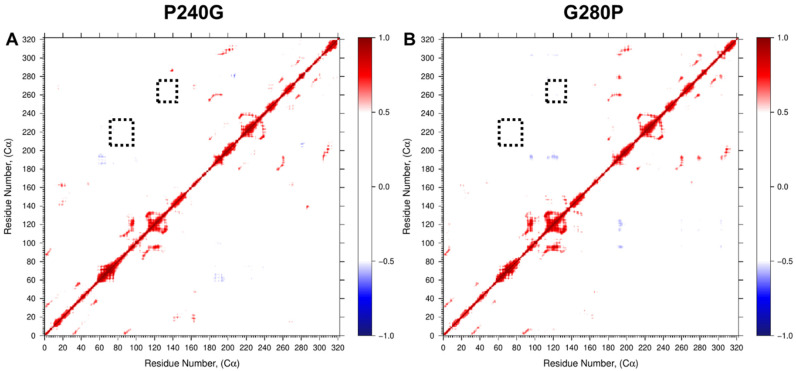
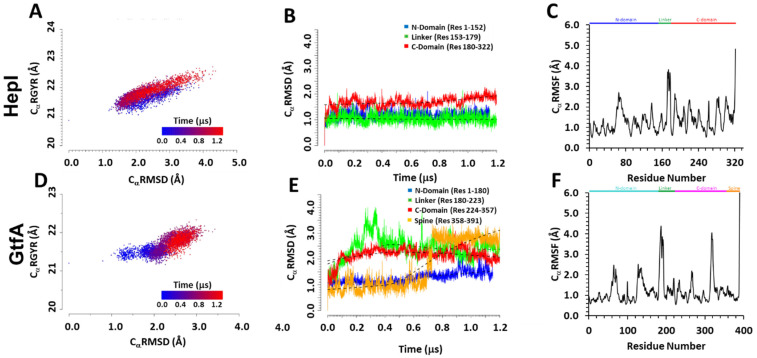
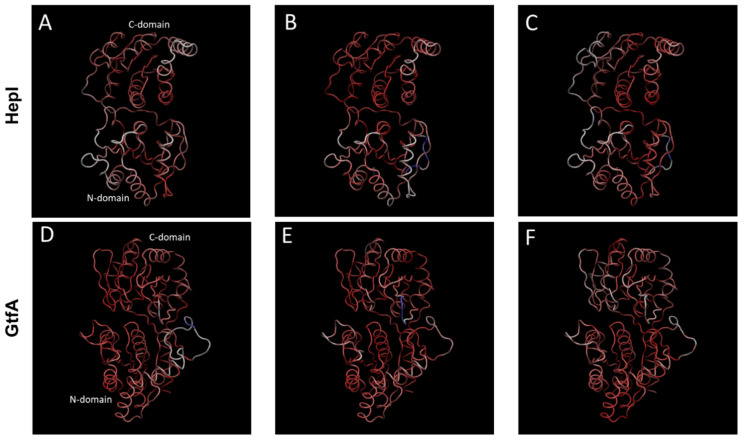
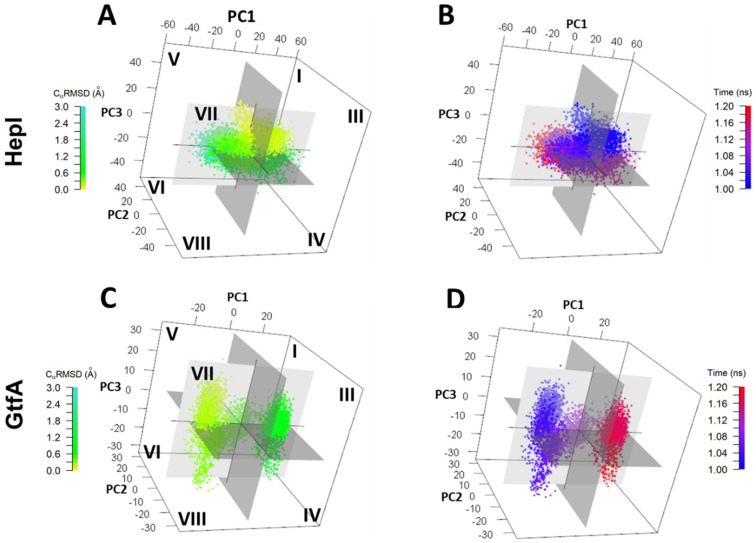
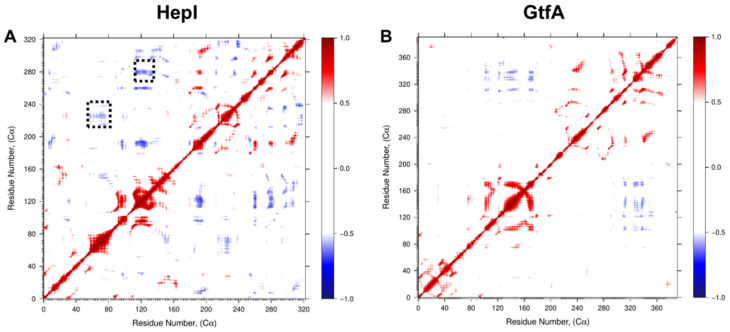
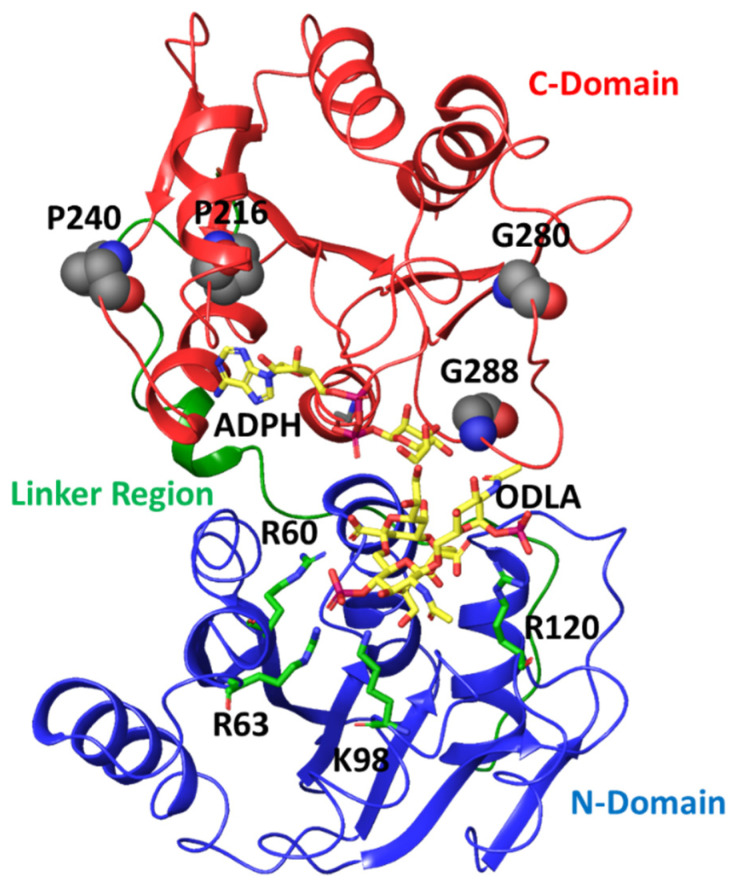
It has long been understood that some proteins undergo conformational transitions en route to the Michaelis Complex to allow chemistry. Examination of crystal structures of glycosyltransferase enzymes in the GT-B structural class reveals that the presence of ligand in the active site triggers an open-to-closed conformation transition, necessary for their catalytic functions. Herein, we describe microsecond molecular dynamics simulations of two distantly related glycosyltransferases that are part of the GT-B structural superfamily, HepI and GtfA. Simulations were performed using the open and closed conformations of these unbound proteins, respectively, and we sought to identify the major dynamical modes and communication networks that interconnect the open and closed structures. We provide the first reported evidence within the scope of our simulation parameters that the interconversion between open and closed conformations is a hierarchical multistep process which can be a conserved feature of enzymes of the same structural superfamily. Each of these motions involves of a collection of smaller molecular reorientations distributed across both domains, highlighting the complexities of protein dynamic involved in the interconversion process. Additionally, dynamic cross-correlation analysis was employed to explore the potential effect of distal residues on the catalytic efficiency of HepI. Multiple distal nonionizable residues of the C-terminal domain exhibit motions anticorrelated to positively charged residues in the active site in the -terminal domain involved in substrate binding. Mutations of these residues resulted in a reduction in negatively correlated motions and an altered enzymatic efficiency that is dominated by lower K values with effectively unchanged. The findings suggest that residues with opposing conformational motions involved in the opening and closing of the bidomain HepI protein can allosterically alter the population and conformation of the "closed" state, essential to the formation of the Michaelis complex. The stabilization effects of these mutations likely equally influence the energetics of both the ground state and the transition state of the catalytic reaction, leading to the unaltered . Our study provides new insights into the role of conformational dynamics in glycosyltransferase's function and new modality to modulate enzymatic efficiency.
长期以来,人们一直认为一些蛋白质在通往迈克尔复合物的过程中会发生构象转变,以允许化学反应发生。对 GT-B 结构类别的糖基转移酶酶的晶体结构的研究表明,配体在活性部位的存在触发了开-闭构象转变,这对于它们的催化功能是必要的。在此,我们描述了两个属于 GT-B 结构超家族的远缘糖基转移酶的微秒分子动力学模拟,即 HepI 和 GtfA。模拟分别使用这些未结合蛋白的开放和闭合构象进行,我们试图确定将开放和闭合结构相互连接的主要动力学模式和通信网络。我们提供了在我们的模拟参数范围内首次报道的证据,表明开-闭构象之间的转换是一个层次化的多步骤过程,这可能是同一结构超家族酶的保守特征。这些运动中的每一个都涉及到跨越两个结构域的一组较小的分子重新取向,突出了蛋白质动态相互转换过程中所涉及的复杂性。此外,动态互相关分析被用来探索远端残基对 HepI 催化效率的潜在影响。C 末端结构域的多个非电离远端残基表现出与活性部位带正电荷的残基的反向运动,这些残基参与了底物结合。这些残基的突变导致负相关运动减少,并且酶效率改变,主要是由于 K 值降低,而 基本上不变。研究结果表明,涉及 HepI 双域蛋白打开和关闭的构象相反的残基可以变构改变“闭合”状态的种群和构象,这对迈克尔复合物的形成至关重要。这些突变的稳定作用可能同样影响催化反应的基态和过渡态的能量学,导致 不变。我们的研究为构象动力学在糖基转移酶功能中的作用提供了新的见解,并为调节酶效率提供了新的模式。