Department of Medical Genetics, Institute of Clinical Medicine, Faculty of Medicine, University of Oslo and Oslo University Hospital, Oslo, Norway.
Department of Cancer Genetics, Institute for Cancer Research, Oslo University Hospital, 0310, Oslo, Norway.
Genome Med. 2021 Apr 29;13(1):72. doi: 10.1186/s13073-021-00880-4.
Abnormal DNA methylation is observed as an early event in breast carcinogenesis. However, how such alterations arise is still poorly understood. microRNAs (miRNAs) regulate gene expression at the post-transcriptional level and play key roles in various biological processes. Here, we integrate miRNA expression and DNA methylation at CpGs to study how miRNAs may affect the breast cancer methylome and how DNA methylation may regulate miRNA expression.
miRNA expression and DNA methylation data from two breast cancer cohorts, Oslo2 (n = 297) and The Cancer Genome Atlas (n = 439), were integrated through a correlation approach that we term miRNA-methylation Quantitative Trait Loci (mimQTL) analysis. Hierarchical clustering was used to identify clusters of miRNAs and CpGs that were further characterized through analysis of mRNA/protein expression, clinicopathological features, in silico deconvolution, chromatin state and accessibility, transcription factor binding, and long-range interaction data.
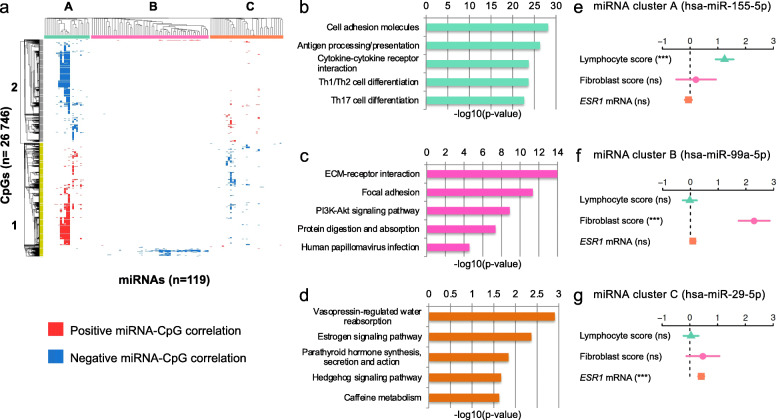
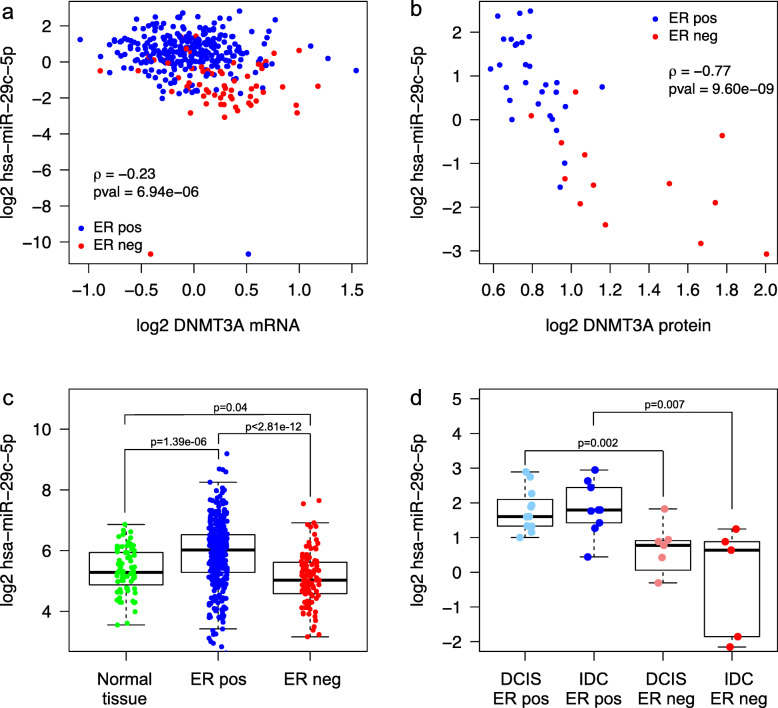
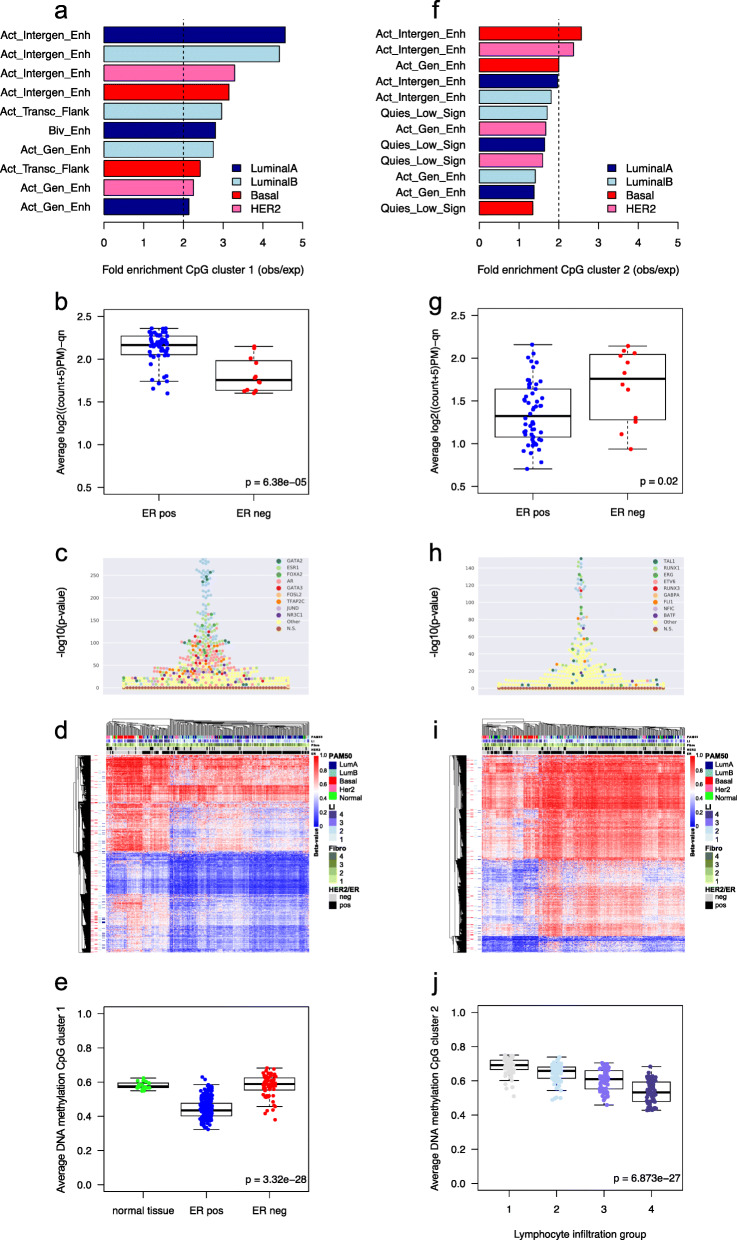
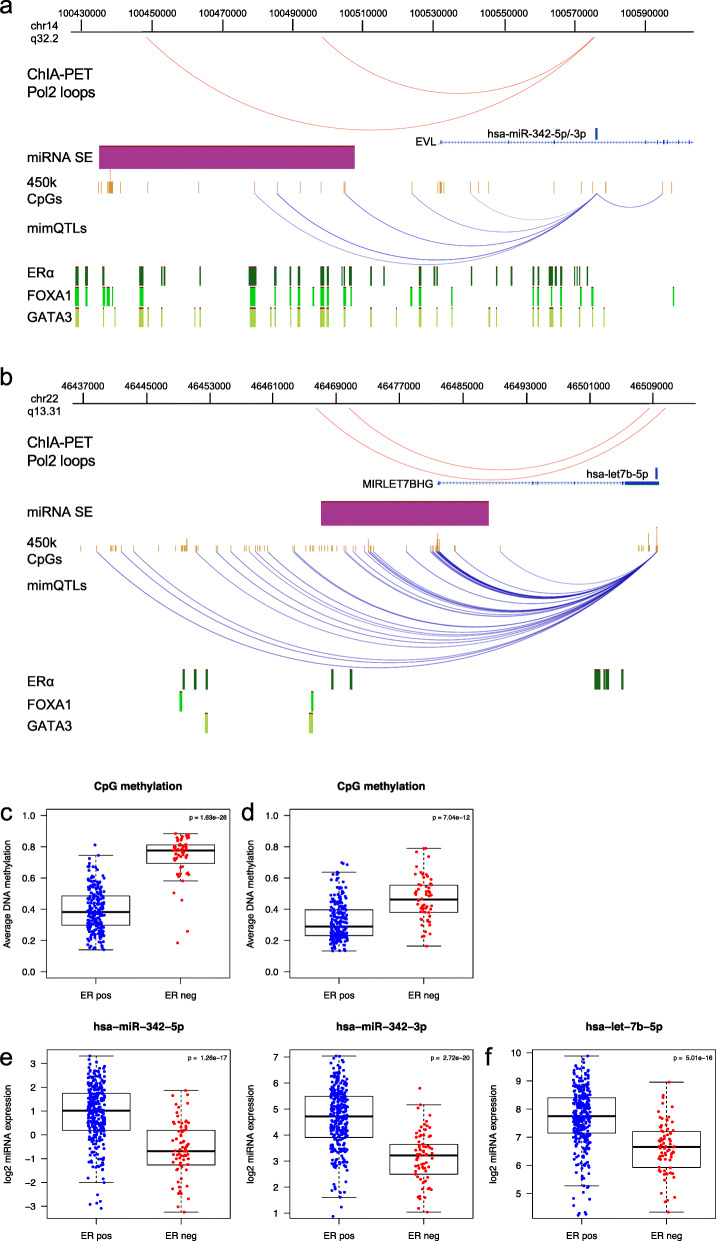
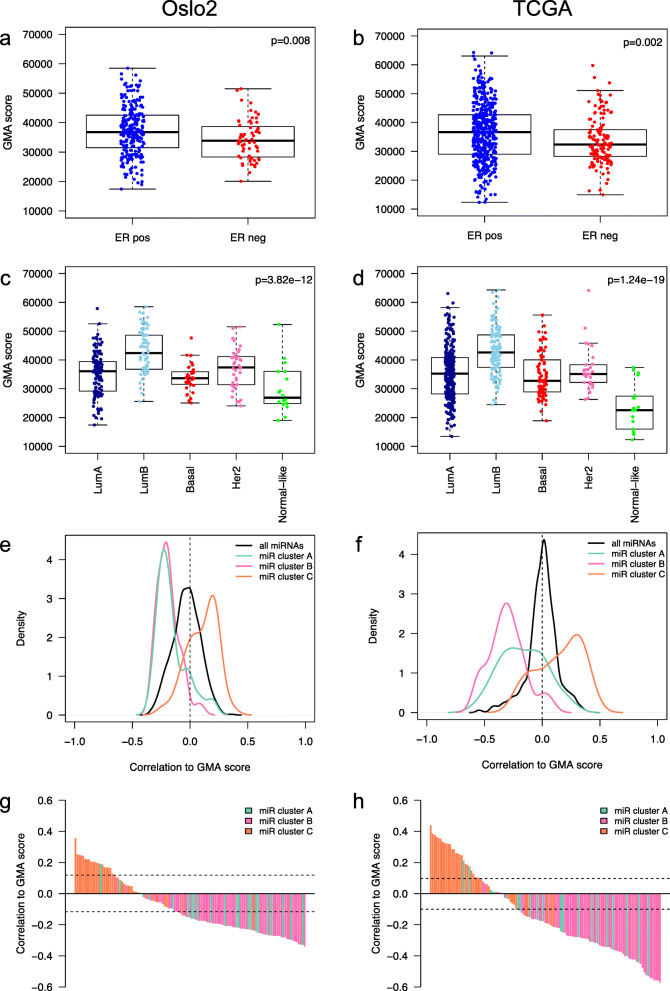
Clustering of the significant mimQTLs identified distinct groups of miRNAs and CpGs that reflect important biological processes associated with breast cancer pathogenesis. Notably, two major miRNA clusters were related to immune or fibroblast infiltration, hence identifying miRNAs associated with cells of the tumor microenvironment, while another large cluster was related to estrogen receptor (ER) signaling. Studying the chromatin landscape surrounding CpGs associated with the estrogen signaling cluster, we found that miRNAs from this cluster are likely to be regulated through DNA methylation of enhancers bound by FOXA1, GATA2, and ER-alpha. Further, at the hub of the estrogen cluster, we identified hsa-miR-29c-5p as negatively correlated with the mRNA and protein expression of DNA methyltransferase DNMT3A, a key enzyme regulating DNA methylation. We found deregulation of hsa-miR-29c-5p already present in pre-invasive breast lesions and postulate that hsa-miR-29c-5p may trigger early event abnormal DNA methylation in ER-positive breast cancer.
We describe how miRNA expression and DNA methylation interact and associate with distinct breast cancer phenotypes.
异常的 DNA 甲基化在乳腺癌发生的早期就被观察到。然而,这些改变是如何产生的仍然知之甚少。microRNAs(miRNAs)在转录后水平上调节基因表达,在各种生物学过程中发挥关键作用。在这里,我们整合 miRNA 的表达和 CpG 上的 DNA 甲基化,研究 miRNA 如何影响乳腺癌甲基组,以及 DNA 甲基化如何调节 miRNA 的表达。
通过我们称之为 miRNA-甲基化定量性状基因座(mimQTL)分析的相关方法,整合了来自两个乳腺癌队列(Oslo2,n=297;癌症基因组图谱,n=439)的 miRNA 表达和 DNA 甲基化数据。使用层次聚类来识别 miRNA 和 CpG 的聚类,这些聚类进一步通过分析 mRNA/蛋白质表达、临床病理特征、计算去卷积、染色质状态和可及性、转录因子结合和长程相互作用数据进行表征。
显著 mimQTL 的聚类确定了 miRNA 和 CpG 的不同簇,这些簇反映了与乳腺癌发病机制相关的重要生物学过程。值得注意的是,两个主要的 miRNA 簇与免疫或成纤维细胞浸润有关,因此鉴定了与肿瘤微环境细胞相关的 miRNA,而另一个大的簇与雌激素受体(ER)信号有关。研究与雌激素信号簇相关的 CpG 周围的染色质景观,我们发现该簇中的 miRNA 可能通过 FOXA1、GATA2 和 ER-α 结合的增强子的 DNA 甲基化来调节。此外,在雌激素簇的中心,我们鉴定出 hsa-miR-29c-5p 与 DNA 甲基转移酶 DNMT3A 的 mRNA 和蛋白质表达呈负相关,DNMT3A 是调节 DNA 甲基化的关键酶。我们发现 hsa-miR-29c-5p 的失调已经存在于侵袭前的乳腺病变中,并推测 hsa-miR-29c-5p 可能引发 ER 阳性乳腺癌中早期异常的 DNA 甲基化。
我们描述了 miRNA 表达和 DNA 甲基化如何相互作用,并与不同的乳腺癌表型相关联。