Watanabe Narumi, Ohnuki Yuuto, Sakakibara Yasubumi
Department of Biosciences and Informatics, Keio University, 3-14-1 Hiyoshi, Kohoku-ku, Yokohama, Kanagawa, 223-8522, Japan.
J Cheminform. 2021 May 1;13(1):36. doi: 10.1186/s13321-021-00513-3.
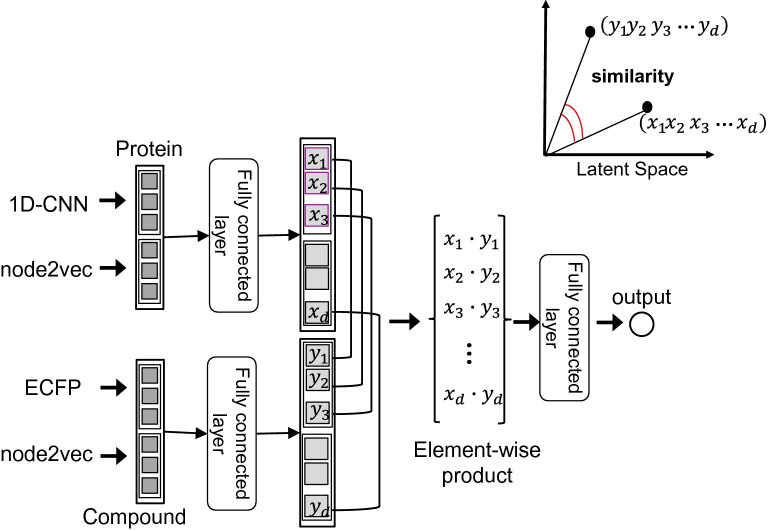
Virtual screening, which can computationally predict the presence or absence of protein-compound interactions, has attracted attention as a large-scale, low-cost, and short-term search method for seed compounds. Existing machine learning methods for predicting protein-compound interactions are largely divided into those based on molecular structure data and those based on network data. The former utilize information on proteins and compounds, such as amino acid sequences and chemical structures; the latter rely on interaction network data, such as protein-protein interactions and compound-compound interactions. However, there have been few attempts to combine both types of data in molecular information and interaction networks.
We developed a deep learning-based method that integrates protein features, compound features, and multiple types of interactome data to predict protein-compound interactions. We designed three benchmark datasets with different difficulties and applied them to evaluate the prediction method. The performance evaluations show that our deep learning framework for integrating molecular structure data and interactome data outperforms state-of-the-art machine learning methods for protein-compound interaction prediction tasks. The performance improvement is statistically significant according to the Wilcoxon signed-rank test. This finding reveals that the multi-interactome data captures perspectives other than amino acid sequence homology and chemical structure similarity and that both types of data synergistically improve the prediction accuracy. Furthermore, experiments on the three benchmark datasets show that our method is more robust than existing methods in accurately predicting interactions between proteins and compounds that are unseen in training samples.
虚拟筛选能够通过计算预测蛋白质与化合物之间相互作用的存在与否,作为一种大规模、低成本且短期的先导化合物搜索方法而备受关注。现有的用于预测蛋白质与化合物相互作用的机器学习方法主要分为基于分子结构数据的方法和基于网络数据的方法。前者利用蛋白质和化合物的信息,如氨基酸序列和化学结构;后者则依赖于相互作用网络数据,如蛋白质 - 蛋白质相互作用和化合物 - 化合物相互作用。然而,在分子信息和相互作用网络中结合这两种类型的数据的尝试却很少。
我们开发了一种基于深度学习的方法,该方法整合了蛋白质特征、化合物特征和多种类型的相互作用组数据来预测蛋白质与化合物之间的相互作用。我们设计了三个具有不同难度的基准数据集,并将其应用于评估预测方法。性能评估表明,我们用于整合分子结构数据和相互作用组数据的深度学习框架在蛋白质 - 化合物相互作用预测任务上优于现有的机器学习方法。根据威尔科克森符号秩检验,性能提升具有统计学意义。这一发现揭示了多相互作用组数据捕捉到了除氨基酸序列同源性和化学结构相似性之外的其他视角,并且这两种类型的数据协同提高了预测准确性。此外,在三个基准数据集上的实验表明,我们的方法在准确预测训练样本中未出现的蛋白质与化合物之间的相互作用方面比现有方法更稳健。