Choueiry Fouad, Singh Satishkumar, Sircar Anuvrat, Laliotis Georgios, Sun Xiaowei, Chavdoula Evangelia, Zhang Shiqi, Helmig-Mason JoBeth, Hart Amber, Epperla Narendranath, Tsichlis Philip, Baiocchi Robert, Alinari Lapo, Zhu Jiangjiang, Sehgal Lalit
Department of Human Sciences, The Ohio State University, Columbus, OH 43210, USA.
Division of Hematology, Department of Internal Medicine, The Ohio State University, Columbus, OH 43210, USA.
Cancers (Basel). 2021 Apr 29;13(9):2146. doi: 10.3390/cancers13092146.
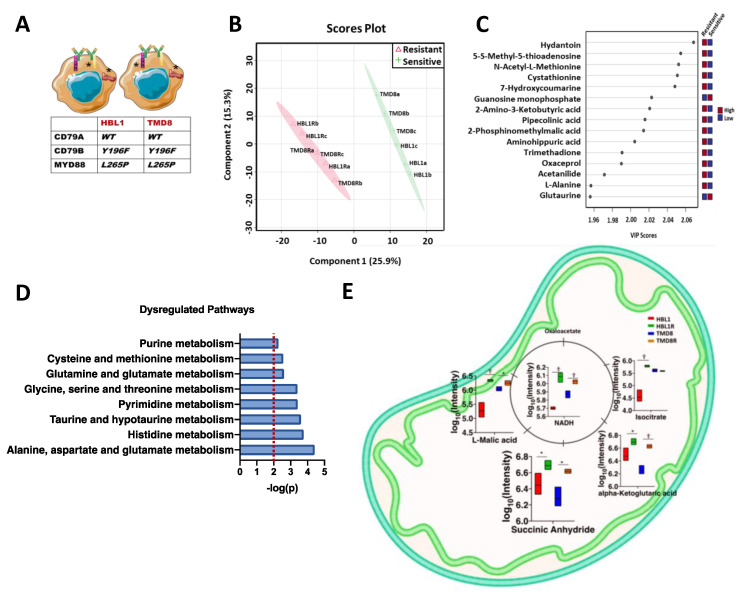
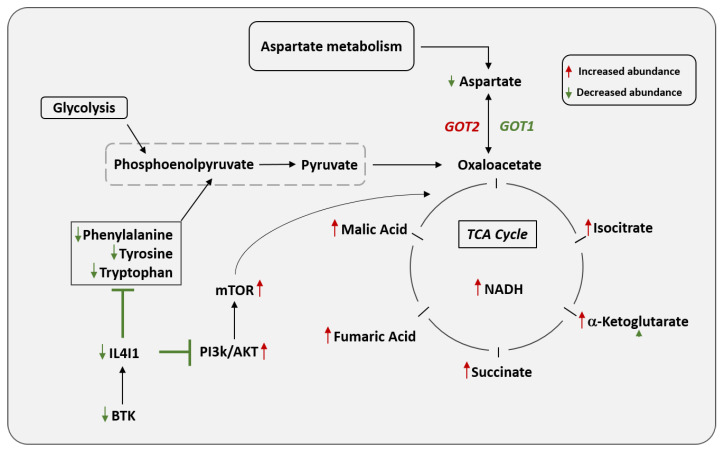
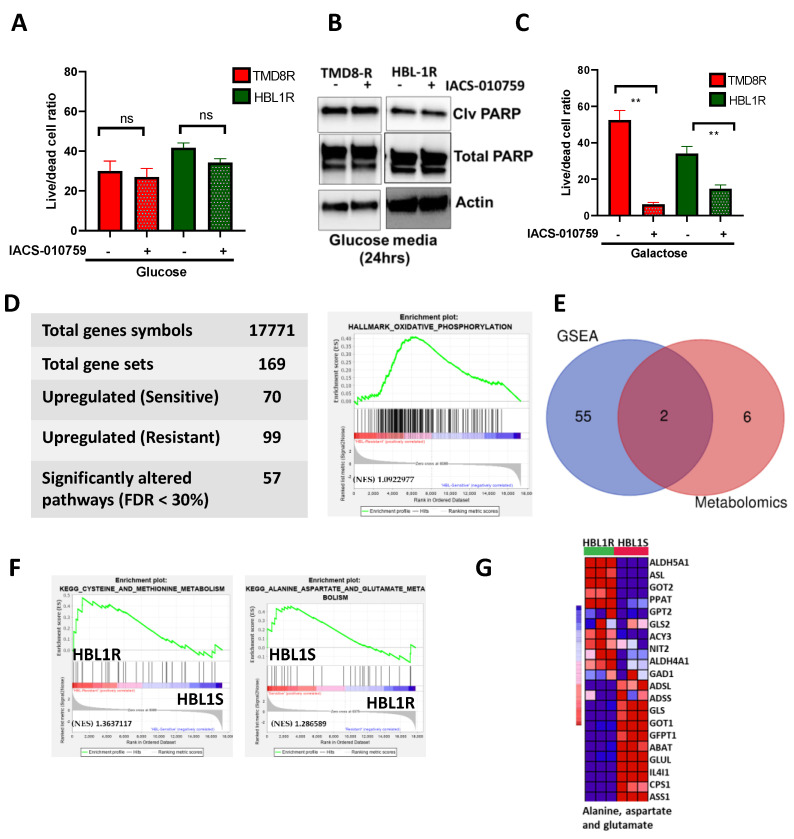
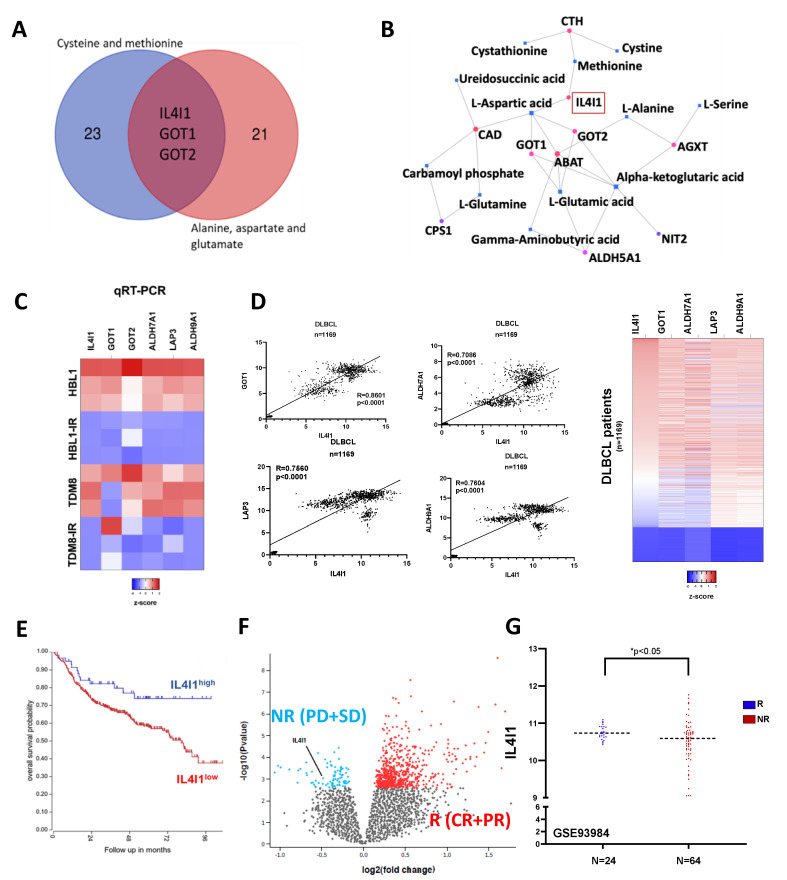
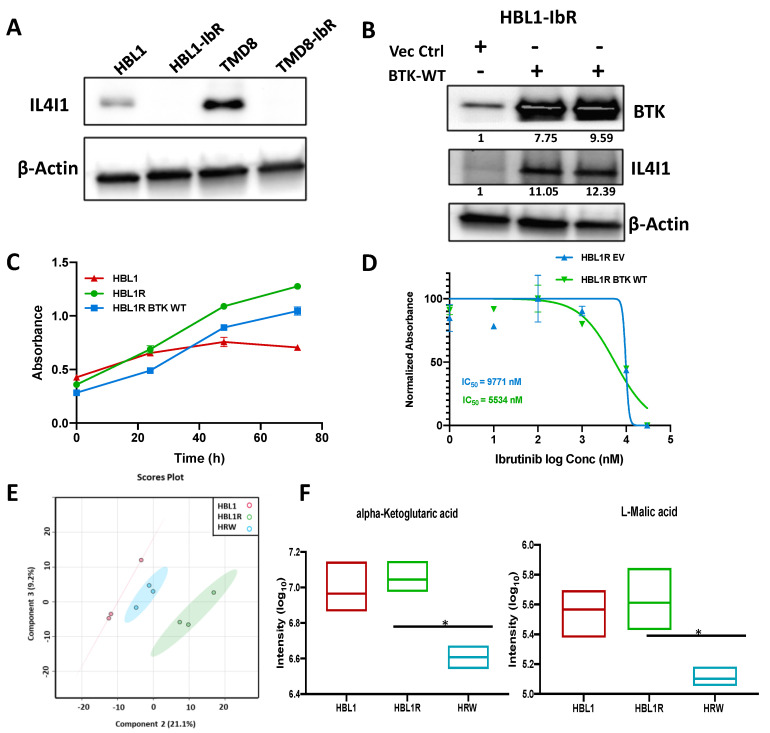
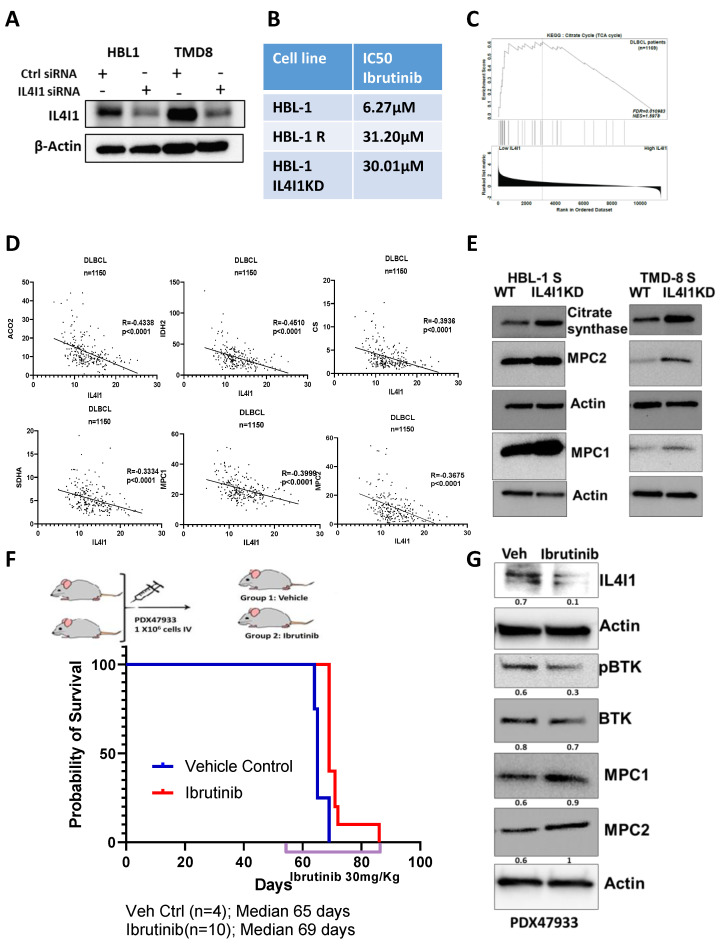
Diffuse large B-cell lymphoma (DLBCL) is the most common non-Hodgkin lymphoma (NHL). B-cell NHLs rely on Bruton's tyrosine kinase (BTK) mediated B-cell receptor signaling for survival and disease progression. However, they are often resistant to BTK inhibitors or soon acquire resistance after drug exposure resulting in the drug-tolerant form. The drug-tolerant clones proliferate faster, have increased metabolic activity, and shift to oxidative phosphorylation; however, how this metabolic programming occurs in the drug-resistant tumor is poorly understood. In this study, we explored for the first time the metabolic regulators of ibrutinib-resistant activated B-cell (ABC) DLBCL using a multi-omics analysis that integrated metabolomics (using high-resolution mass spectrometry) and transcriptomic (gene expression analysis). Overlay of the unbiased statistical analyses, genetic perturbation, and pharmaceutical inhibition was further used to identify the key players contributing to the metabolic reprogramming of the drug-resistant clone. Gene-metabolite integration revealed interleukin four induced 1 (IL4I1) at the crosstalk of two significantly altered metabolic pathways involved in producing various amino acids. We showed for the first time that drug-resistant clones undergo metabolic reprogramming towards oxidative phosphorylation and are modulated via the BTK-PI3K-AKT-IL4I1 axis. Our report shows how these cells become dependent on PI3K/AKT signaling for survival after acquiring ibrutinib resistance and shift to sustained oxidative phosphorylation; additionally, we outline the compensatory pathway that might regulate this metabolic reprogramming in the drug-resistant cells. These findings from our unbiased analyses highlight the role of metabolic reprogramming during drug resistance development. Our work demonstrates that a multi-omics approach can be a robust and impartial strategy to uncover genes and pathways that drive metabolic deregulation in cancer cells.
弥漫性大B细胞淋巴瘤(DLBCL)是最常见的非霍奇金淋巴瘤(NHL)。B细胞NHL依赖布鲁顿酪氨酸激酶(BTK)介导的B细胞受体信号传导来维持生存和疾病进展。然而,它们通常对BTK抑制剂耐药,或在药物暴露后很快产生耐药性,从而形成耐药形式。耐药克隆增殖更快,代谢活性增加,并转向氧化磷酸化;然而,这种代谢重编程在耐药肿瘤中是如何发生的,目前还知之甚少。在本研究中,我们首次使用整合代谢组学(使用高分辨率质谱)和转录组学(基因表达分析)的多组学分析方法,探索了依鲁替尼耐药的活化B细胞(ABC)DLBCL的代谢调节因子。通过无偏统计分析、基因扰动和药物抑制的叠加,进一步确定了导致耐药克隆代谢重编程的关键因素。基因-代谢物整合揭示了白细胞介素4诱导1(IL4I1)在参与产生各种氨基酸的两条显著改变的代谢途径的交汇处。我们首次表明,耐药克隆向氧化磷酸化方向进行代谢重编程,并通过BTK-PI3K-AKT-IL4I1轴进行调节。我们的报告显示了这些细胞在获得依鲁替尼耐药性后如何依赖PI3K/AKT信号传导来维持生存,并转向持续的氧化磷酸化;此外,我们概述了可能调节耐药细胞中这种代谢重编程的补偿途径。我们无偏分析得出的这些发现突出了代谢重编程在耐药发展过程中的作用。我们的工作表明,多组学方法可以是一种强大而公正的策略,用于揭示驱动癌细胞代谢失调的基因和途径。