SINTEF DIGITAL, Forskningsveien 1, 0373, Oslo, Norway.
Department of Mathematical Sciences, Norwegian University of Science and Technology, A. Getz vei 1, 7491, Trondheim, Norway.
BMC Bioinformatics. 2021 May 4;22(1):230. doi: 10.1186/s12859-021-04041-7.
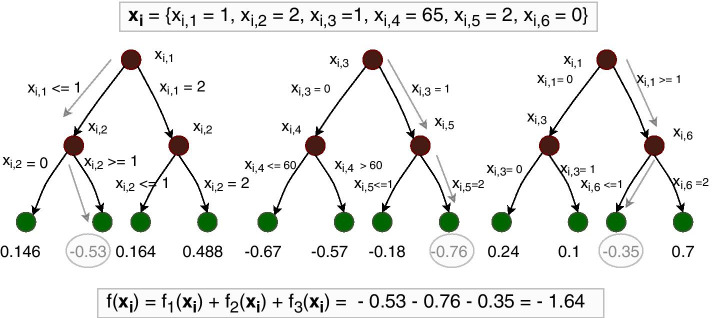
The identification of gene-gene and gene-environment interactions in genome-wide association studies is challenging due to the unknown nature of the interactions and the overwhelmingly large number of possible combinations. Parametric regression models are suitable to look for prespecified interactions. Nonparametric models such as tree ensemble models, with the ability to detect any unspecified interaction, have previously been difficult to interpret. However, with the development of methods for model explainability, it is now possible to interpret tree ensemble models efficiently and with a strong theoretical basis.
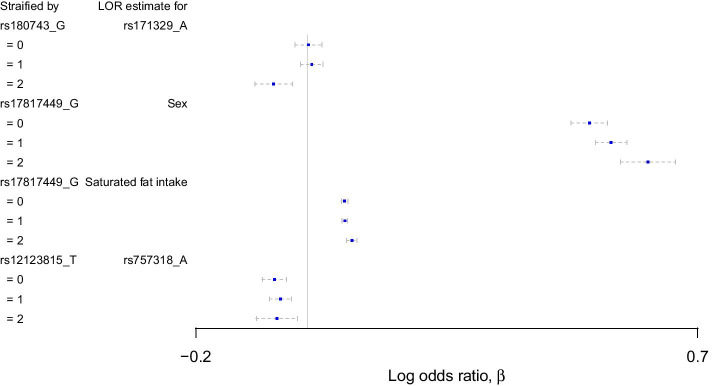
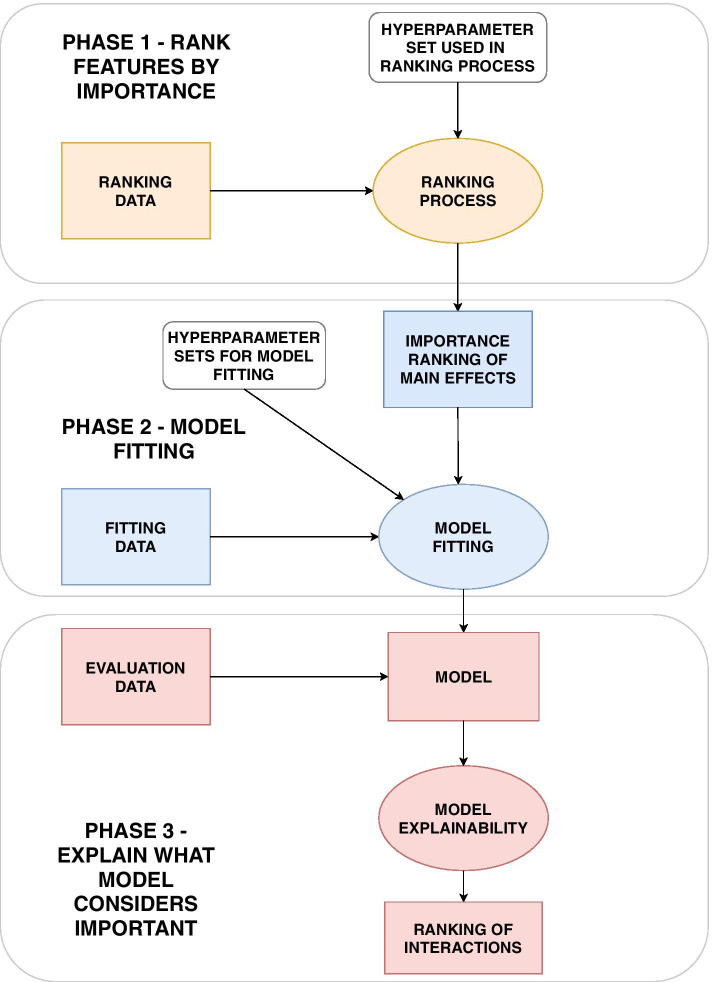
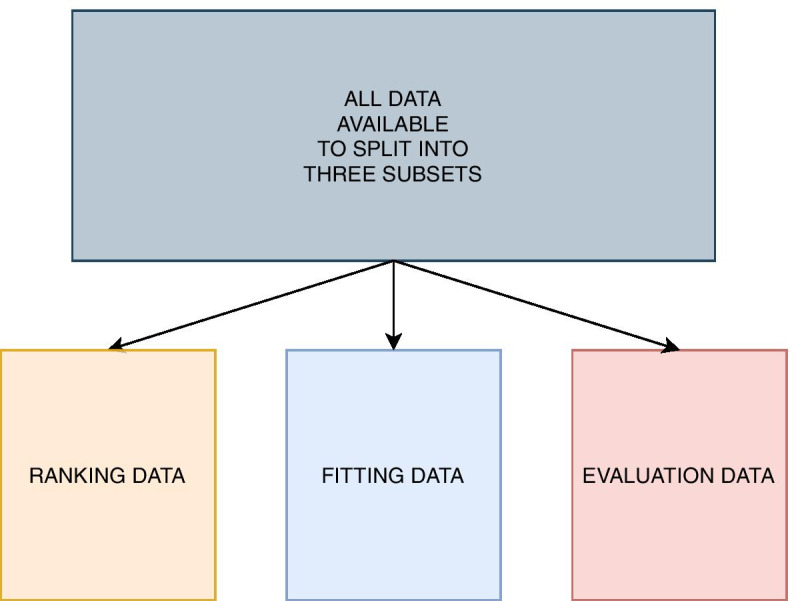
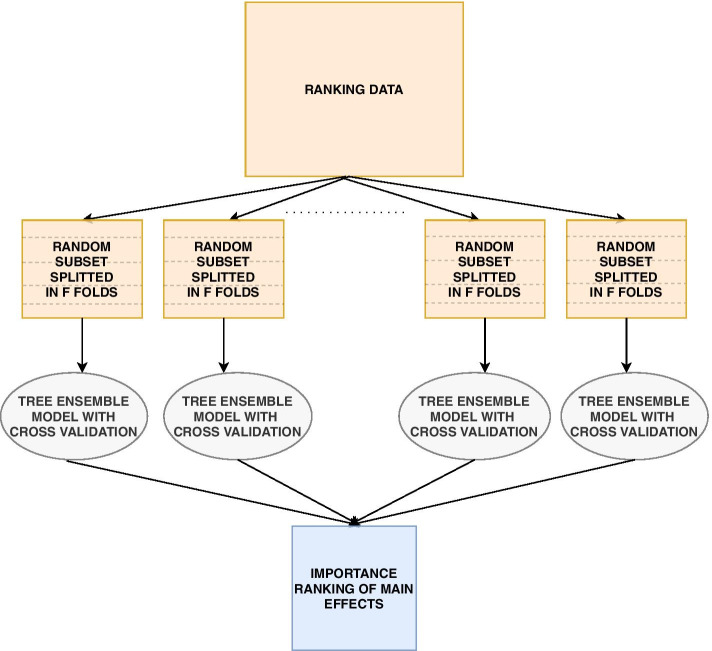
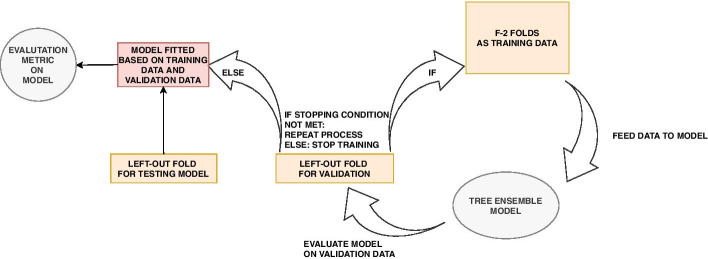
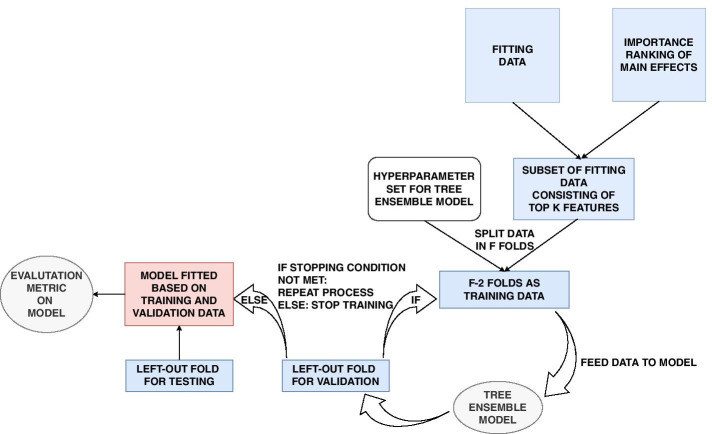
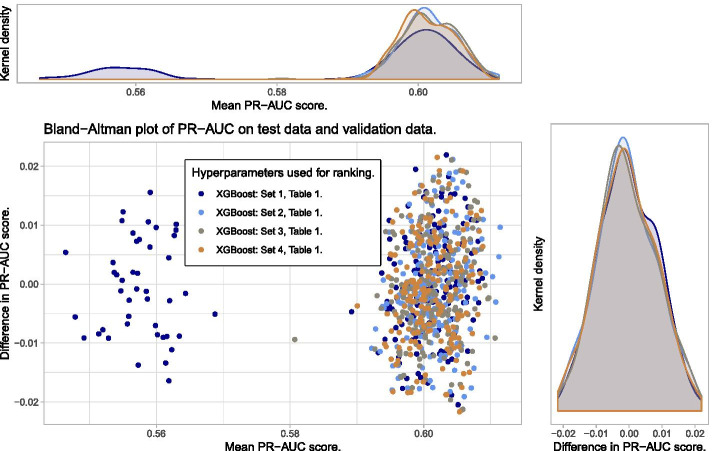
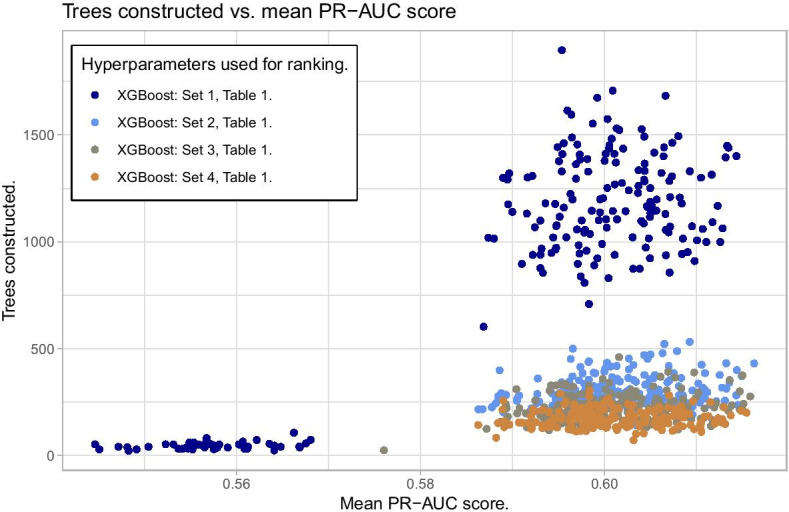
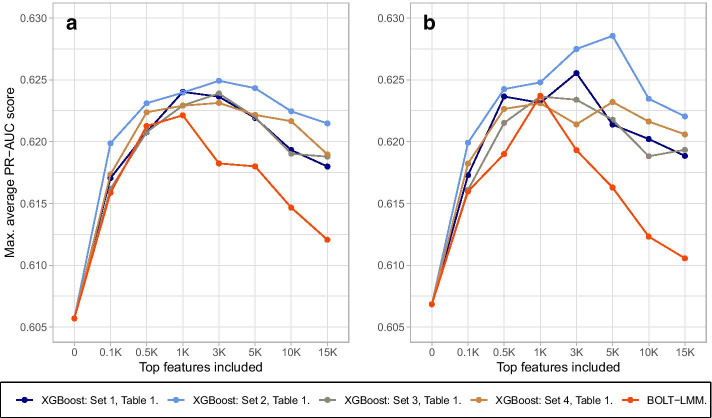
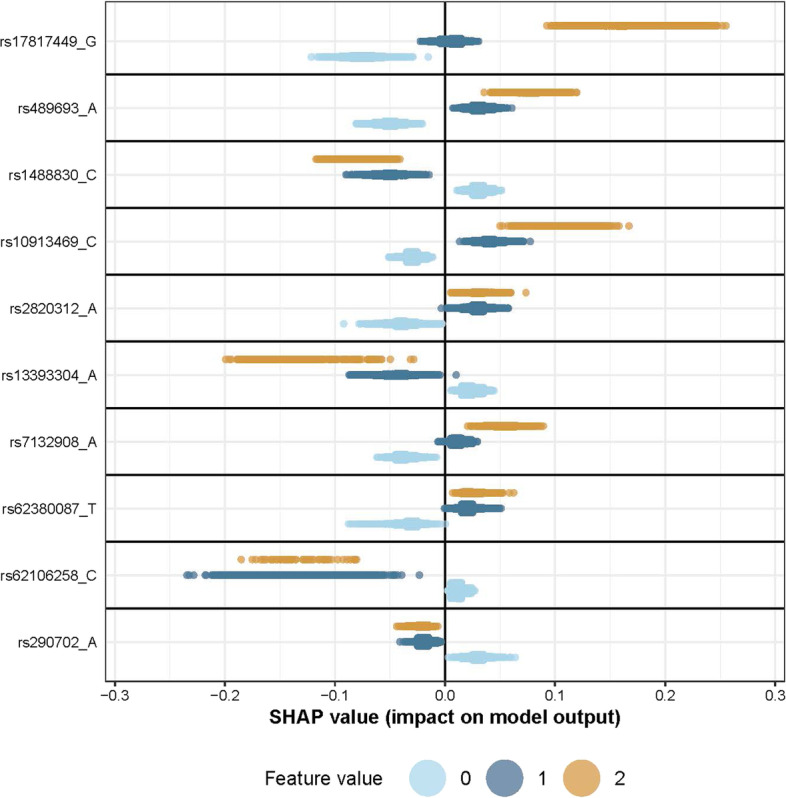
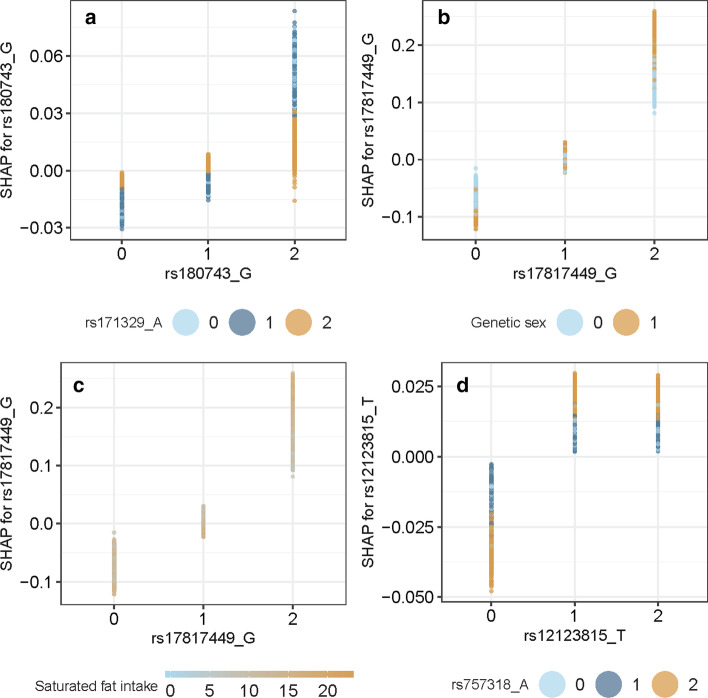
We propose a tree ensemble- and SHAP-based method for identifying as well as interpreting potential gene-gene and gene-environment interactions on large-scale biobank data. A set of independent cross-validation runs are used to implicitly investigate the whole genome. We apply and evaluate the method using data from the UK Biobank with obesity as the phenotype. The results are in line with previous research on obesity as we identify top SNPs previously associated with obesity. We further demonstrate how to interpret and visualize interaction candidates.
The new method identifies interaction candidates otherwise not detected with parametric regression models. However, further research is needed to evaluate the uncertainties of these candidates. The method can be applied to large-scale biobanks with high-dimensional data.
由于交互作用的未知性质和可能的组合数量过多,全基因组关联研究中基因-基因和基因-环境交互作用的识别具有挑战性。参数回归模型适合寻找预设的交互作用。树集成模型等非参数模型具有检测任何未指定交互作用的能力,但以前很难解释。然而,随着模型可解释性方法的发展,现在可以有效地解释树集成模型,并具有很强的理论基础。
我们提出了一种基于树集成和 SHAP 的方法,用于在大规模生物库数据中识别和解释潜在的基因-基因和基因-环境交互作用。一组独立的交叉验证运行用于隐式地研究整个基因组。我们使用 UK Biobank 中的肥胖作为表型数据来应用和评估该方法。结果与肥胖的先前研究一致,因为我们确定了之前与肥胖相关的顶级 SNP。我们进一步演示了如何解释和可视化交互作用候选者。
该新方法可以识别参数回归模型无法检测到的交互作用候选者。然而,需要进一步研究来评估这些候选者的不确定性。该方法可以应用于具有高维数据的大规模生物库。