Department of Plant and Microbial Biology, University of Minnesota-Twin Cities, 1445 Gortner Avenue, St. Paul, MN 55108, USA.
Department of Biological Sciences and Institute for Bioinformatics and Evolutionary Studies, University of Idaho, 875 Perimeter Drive MS 3051, Moscow, ID 83844, USA.
Syst Biol. 2021 Dec 16;71(1):190-207. doi: 10.1093/sysbio/syab032.
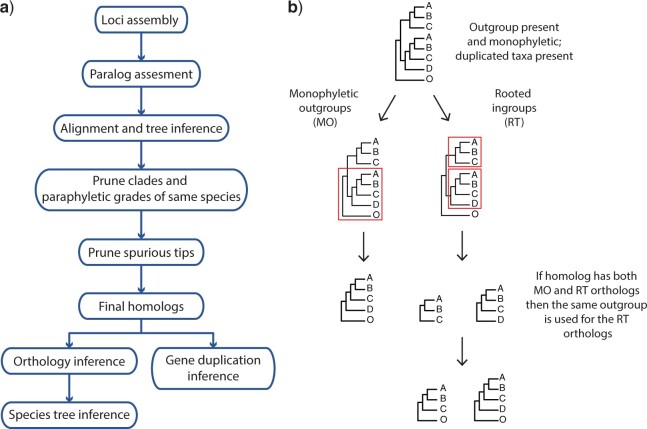
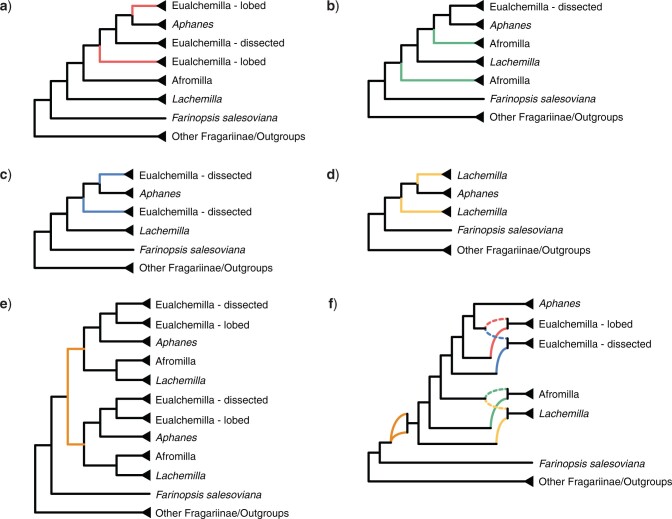
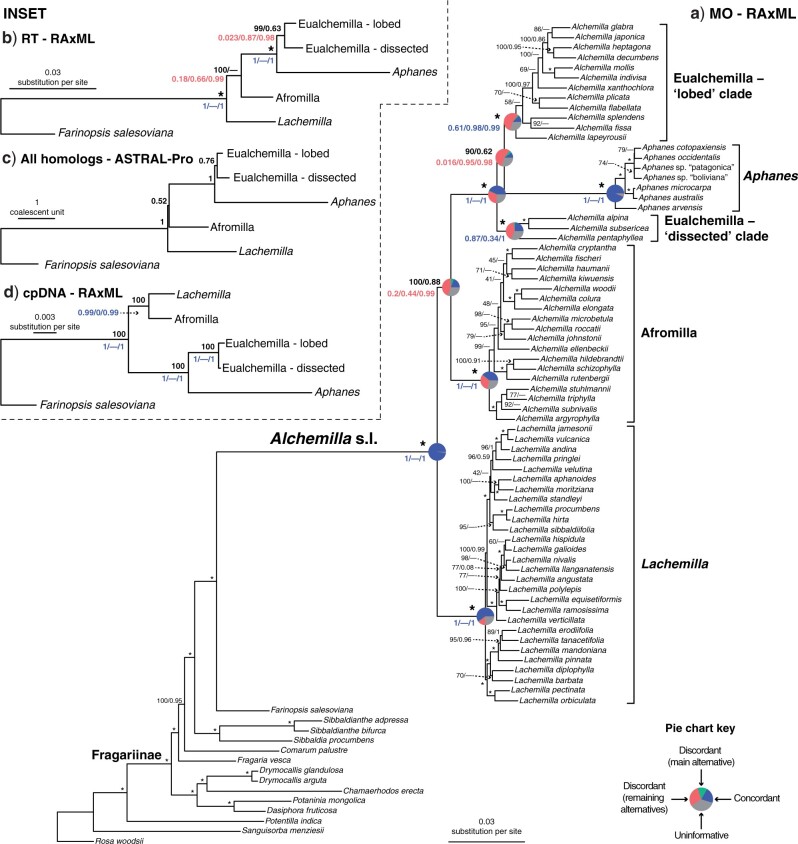
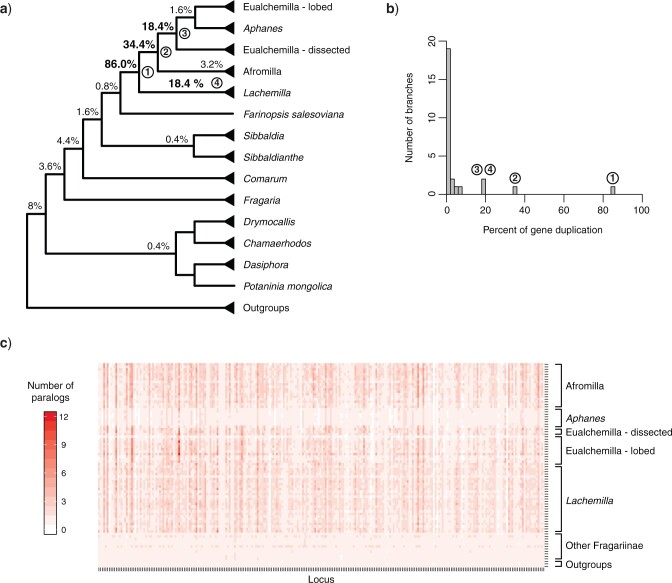
Target enrichment is becoming increasingly popular for phylogenomic studies. Although baits for enrichment are typically designed to target single-copy genes, paralogs are often recovered with increased sequencing depth, sometimes from a significant proportion of loci, especially in groups experiencing whole-genome duplication (WGD) events. Common approaches for processing paralogs in target enrichment data sets include random selection, manual pruning, and mainly, the removal of entire genes that show any evidence of paralogy. These approaches are prone to errors in orthology inference or removing large numbers of genes. By removing entire genes, valuable information that could be used to detect and place WGD events is discarded. Here, we used an automated approach for orthology inference in a target enrichment data set of 68 species of Alchemilla s.l. (Rosaceae), a widely distributed clade of plants primarily from temperate climate regions. Previous molecular phylogenetic studies and chromosome numbers both suggested ancient WGDs in the group. However, both the phylogenetic location and putative parental lineages of these WGD events remain unknown. By taking paralogs into consideration and inferring orthologs from target enrichment data, we identified four nodes in the backbone of Alchemilla s.l. with an elevated proportion of gene duplication. Furthermore, using a gene-tree reconciliation approach, we established the autopolyploid origin of the entire Alchemilla s.l. and the nested allopolyploid origin of four major clades within the group. Here, we showed the utility of automated tree-based orthology inference methods, previously designed for genomic or transcriptomic data sets, to study complex scenarios of polyploidy and reticulate evolution from target enrichment data sets.[Alchemilla; allopolyploidy; autopolyploidy; gene tree discordance; orthology inference; paralogs; Rosaceae; target enrichment; whole genome duplication.].
靶向富集在系统发育基因组学研究中越来越受欢迎。尽管富集的诱饵通常设计用于靶向单拷贝基因,但随着测序深度的增加,通常会从大量基因中回收旁系同源物,尤其是在经历全基因组复制(WGD)事件的群体中。处理靶向富集数据集中外源基因的常见方法包括随机选择、手动修剪,主要是去除任何旁系同源证据的整个基因。这些方法容易在同源性推断或去除大量基因时出错。通过去除整个基因,丢弃了可能用于检测和定位 WGD 事件的有价值信息。在这里,我们使用一种自动化方法来推断 68 种阿尔凯米拉属(蔷薇科)植物的靶向富集数据集的同源性,这是一个广泛分布的植物类群,主要来自温带气候地区。以前的分子系统发育研究和染色体数量都表明该组存在古老的 WGD。然而,这些 WGD 事件的系统发育位置和假定的亲本谱系仍然未知。通过考虑旁系同源物并从靶向富集数据中推断同源物,我们在阿尔凯米拉属的骨干中确定了四个具有较高基因重复比例的节点。此外,使用基因树和解离方法,我们确定了整个阿尔凯米拉属的同源多倍体起源和该组内四个主要分支的嵌套异源多倍体起源。在这里,我们展示了自动化基于树的同源性推断方法的实用性,这些方法以前是为基因组或转录组数据集设计的,用于从靶向富集数据集研究复杂的多倍体和网状进化情景。[阿尔凯米拉属;异源多倍体;同源多倍体;基因树分歧;同源性推断;旁系同源物;蔷薇科;靶向富集;全基因组复制。]。