Li Xuanyi, Xu Yinqiu, Yao Hequan, Lin Kejiang
Department of Medicinal Chemistry, School of Pharmacy, China Pharmaceutical University, 24 Tongjiaxiang, Nanjing, 210009, China.
J Cheminform. 2020 Jun 8;12(1):42. doi: 10.1186/s13321-020-00446-3.
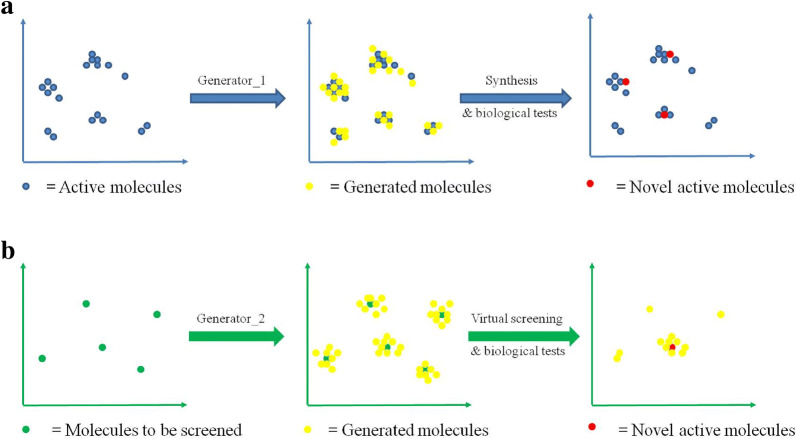
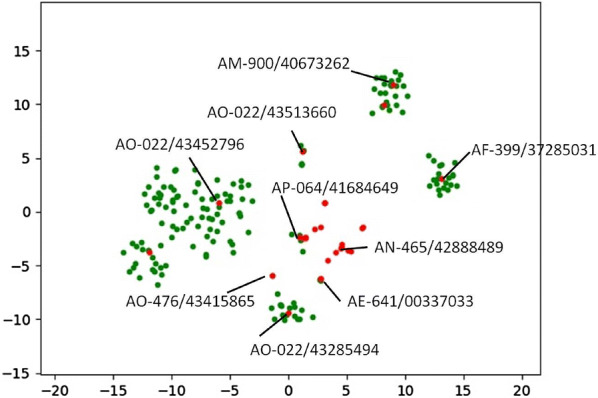
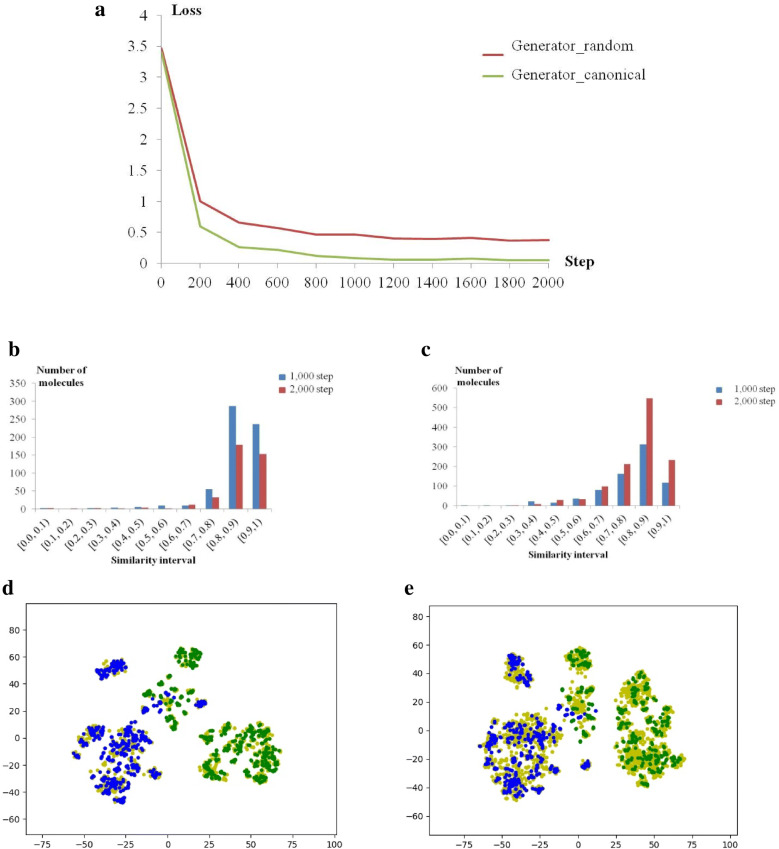
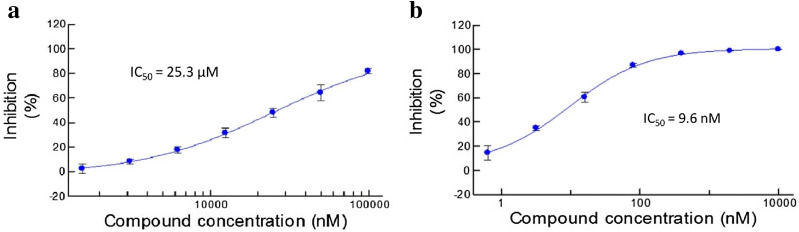
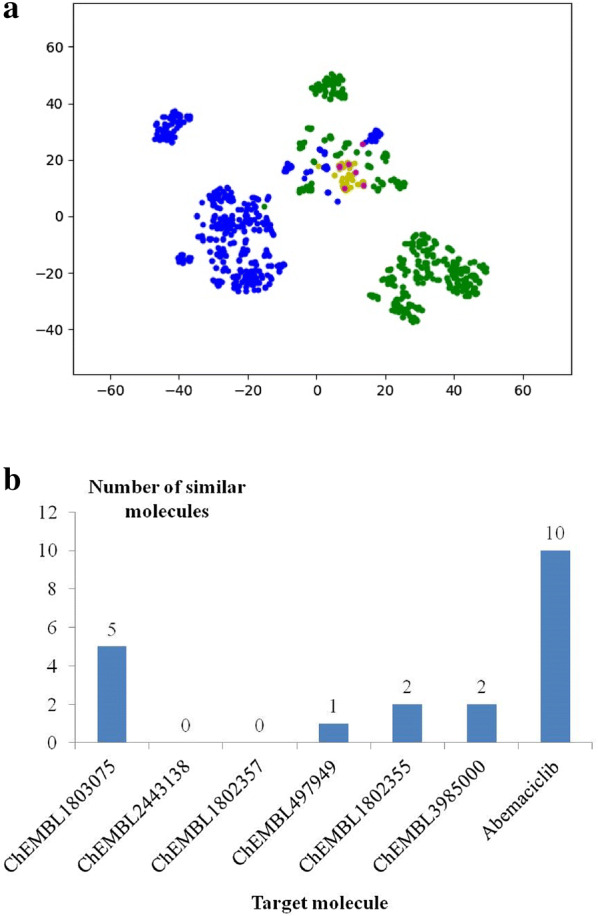
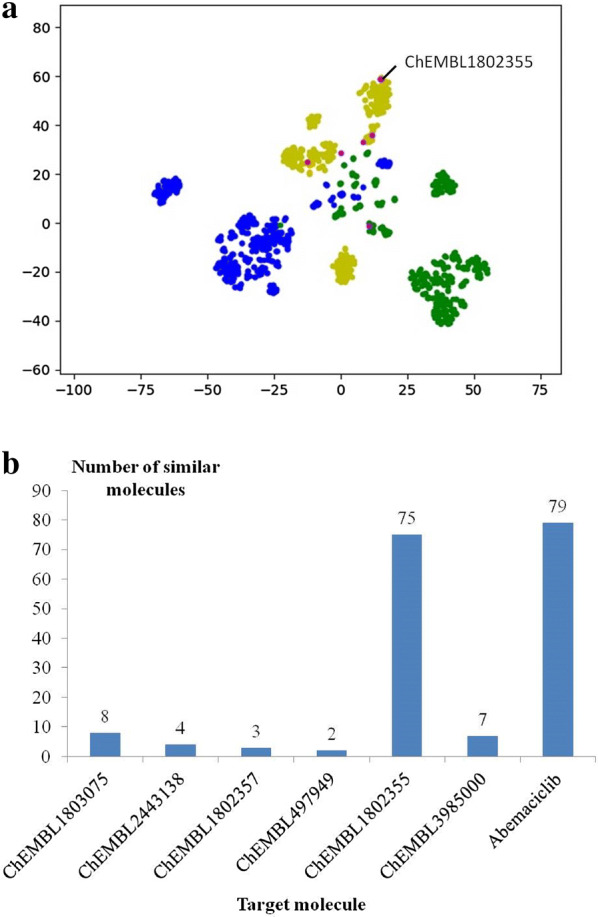
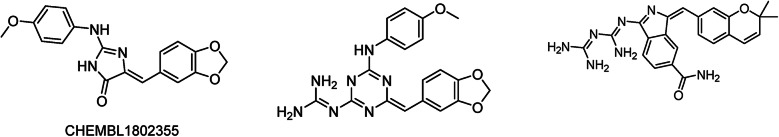
With the rise of artificial intelligence (AI) in drug discovery, de novo molecular generation provides new ways to explore chemical space. However, because de novo molecular generation methods rely on abundant known molecules, generated molecules may have a problem of novelty. Novelty is important in highly competitive areas of medicinal chemistry, such as the discovery of kinase inhibitors. In this study, de novo molecular generation based on recurrent neural networks was applied to discover a new chemical space of kinase inhibitors. During the application, the practicality was evaluated, and new inspiration was found. With the successful discovery of one potent Pim1 inhibitor and two lead compounds that inhibit CDK4, AI-based molecular generation shows potentials in drug discovery and development.
随着人工智能(AI)在药物发现领域的兴起,从头分子生成提供了探索化学空间的新方法。然而,由于从头分子生成方法依赖于大量已知分子,生成的分子可能存在新颖性问题。新颖性在药物化学竞争激烈的领域中很重要,例如激酶抑制剂的发现。在本研究中,基于循环神经网络的从头分子生成被应用于发现激酶抑制剂的新化学空间。在应用过程中,对实用性进行了评估,并获得了新的启发。随着一种有效的Pim1抑制剂和两种抑制CDK4的先导化合物的成功发现,基于人工智能的分子生成在药物发现和开发中显示出潜力。