Molecular Biology, National Measurement Laboratory, LGC, Queens Road, Teddington, TW11 0LY, Middlesex, UK.
Department of Microbiology, Virology and Infection Control, Great Ormond Street Hospital for Children NHS Trust, Great Ormond Street, London, WC1N 3JH, UK.
Sci Rep. 2021 May 19;11(1):10590. doi: 10.1038/s41598-021-89881-2.
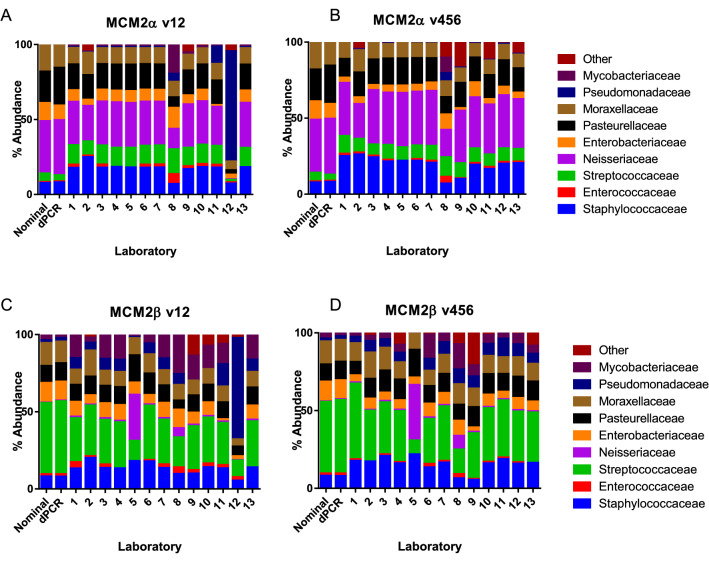
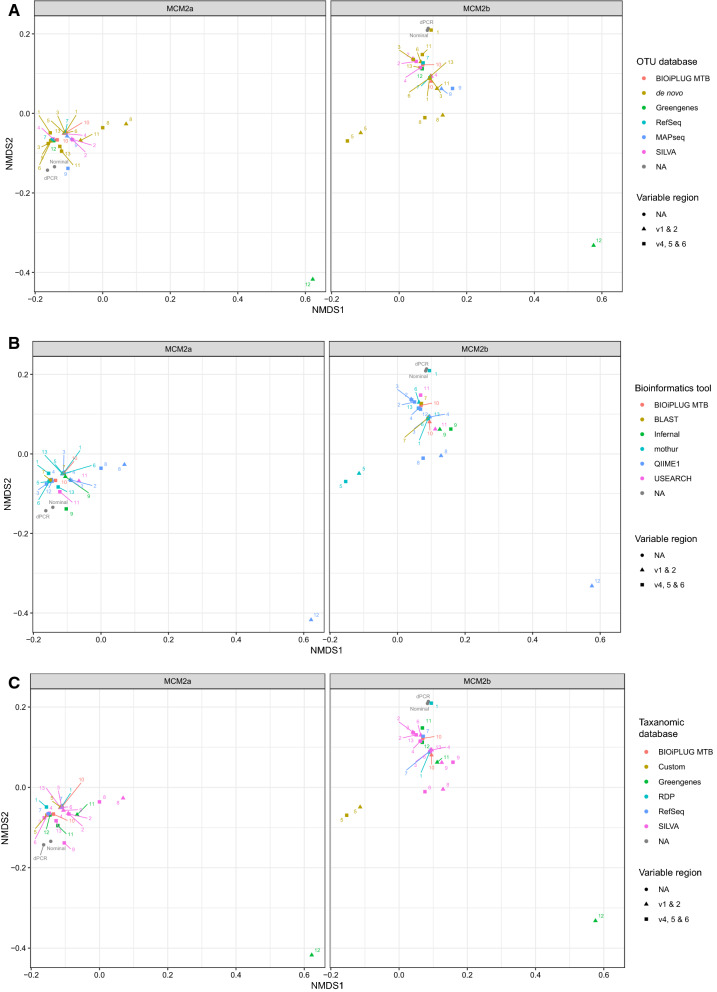
Despite the advent of whole genome metagenomics, targeted approaches (such as 16S rRNA gene amplicon sequencing) continue to be valuable for determining the microbial composition of samples. Amplicon microbiome sequencing can be performed on clinical samples from a normally sterile site to determine the aetiology of an infection (usually single pathogen identification) or samples from more complex niches such as human mucosa or environmental samples where multiple microorganisms need to be identified. The methodologies are frequently applied to determine both presence of micro-organisms and their quantity or relative abundance. There are a number of technical steps required to perform microbial community profiling, many of which may have appreciable precision and bias that impacts final results. In order for these methods to be applied with the greatest accuracy, comparative studies across different laboratories are warranted. In this study we explored the impact of the bioinformatic approaches taken in different laboratories on microbiome assessment using 16S rRNA gene amplicon sequencing results. Data were generated from two mock microbial community samples which were amplified using primer sets spanning five different variable regions of 16S rRNA genes. The PCR-sequencing analysis included three technical repeats of the process to determine the repeatability of their methods. Thirteen laboratories participated in the study, and each analysed the same FASTQ files using their choice of pipeline. This study captured the methods used and the resulting sequence annotation and relative abundance output from bioinformatic analyses. Results were compared to digital PCR assessment of the absolute abundance of each target representing each organism in the mock microbial community samples and also to analyses of shotgun metagenome sequence data. This ring trial demonstrates that the choice of bioinformatic analysis pipeline alone can result in different estimations of the composition of the microbiome when using 16S rRNA gene amplicon sequencing data. The study observed differences in terms of both presence and abundance of organisms and provides a resource for ensuring reproducible pipeline development and application. The observed differences were especially prevalent when using custom databases and applying high stringency operational taxonomic unit (OTU) cut-off limits. In order to apply sequencing approaches with greater accuracy, the impact of different analytical steps needs to be clearly delineated and solutions devised to harmonise microbiome analysis results.
尽管全基因组宏基因组学已经问世,但靶向方法(如 16S rRNA 基因扩增子测序)仍然对于确定样本中的微生物组成非常有价值。扩增子微生物组测序可用于从正常无菌部位的临床样本中进行,以确定感染的病因(通常是单一病原体鉴定),或从更复杂的生态位(如人类粘膜或环境样本)中进行,这些样本需要识别多种微生物。这些方法经常用于确定微生物的存在及其数量或相对丰度。进行微生物群落分析需要许多技术步骤,其中许多步骤可能具有相当大的精度和偏差,从而影响最终结果。为了使这些方法具有最高的准确性,需要在不同的实验室之间进行比较研究。在这项研究中,我们探讨了不同实验室采用的生物信息学方法对使用 16S rRNA 基因扩增子测序结果进行微生物组评估的影响。数据来自两个模拟微生物群落样本,这些样本使用跨越 16S rRNA 基因五个不同可变区的引物组进行扩增。PCR-测序分析包括该过程的三个技术重复,以确定其方法的可重复性。13 个实验室参与了这项研究,每个实验室都使用自己选择的管道分析相同的 FASTQ 文件。本研究捕获了所使用的方法以及生物信息学分析的结果序列注释和相对丰度输出。结果与模拟微生物群落样本中每个目标的绝对丰度的数字 PCR 评估以及对 shotgun 宏基因组序列数据的分析进行了比较。这项环试验表明,仅选择生物信息学分析管道就可以导致使用 16S rRNA 基因扩增子测序数据时微生物组组成的不同估计。该研究观察到了在生物体的存在和丰度方面的差异,并为确保可重复的管道开发和应用提供了资源。当使用自定义数据库和应用高严格性操作分类单元(OTU)截止限值时,观察到的差异尤其普遍。为了更准确地应用测序方法,需要清楚地区分不同分析步骤的影响,并制定解决方案以协调微生物组分析结果。