Zhou You, Xu Bin, Zhou Yi, Liu Jian, Zheng Xiao, Liu Yingting, Deng Haifeng, Liu Ming, Ren Xiubao, Xia Jianchuan, Kong Xiangyin, Huang Tao, Jiang Jingting
Tumor Biological Diagnosis and Treatment Center, The Third Affiliated Hospital of Soochow University, Changzhou, China.
Jiangsu Engineering Research Center for Tumor Immunotherapy, Changzhou, China.
Front Cell Dev Biol. 2021 May 5;9:675438. doi: 10.3389/fcell.2021.675438. eCollection 2021.
With the advent of large-scale molecular profiling, an increasing number of oncogenic drivers contributing to precise medicine and reshaping classification of lung adenocarcinoma (LUAD) have been identified. However, only a minority of patients archived improved outcome under current standard therapies because of the dynamic mutational spectrum, which required expanding susceptible gene libraries. Accumulating evidence has witnessed that understanding gene regulatory networks as well as their changing processes was helpful in identifying core genes which acted as master regulators during carcinogenesis. The present study aimed at identifying key genes with differential correlations between normal and tumor status.
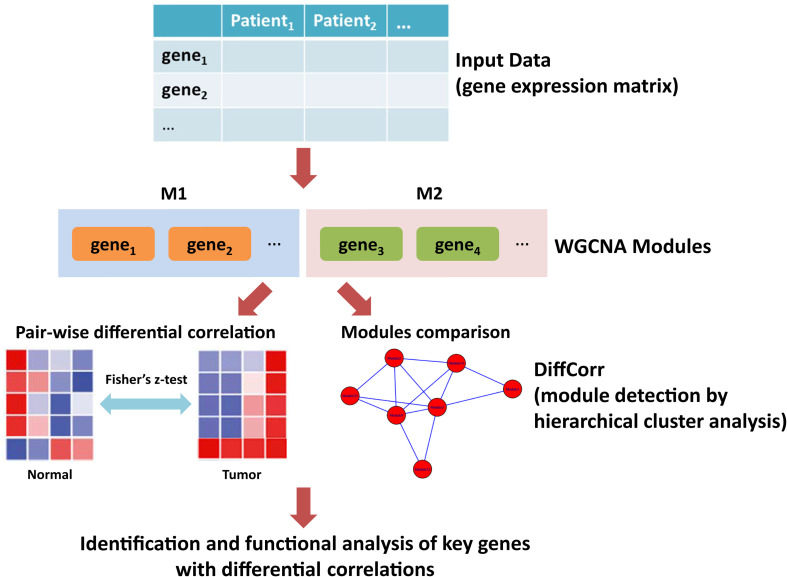
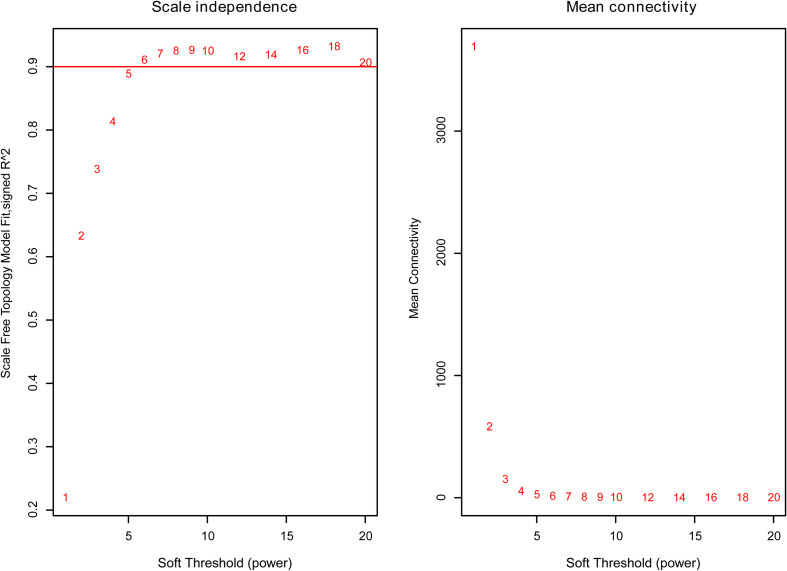
Weighted gene co-expression network analysis (WGCNA) was employed to build a gene interaction network using the expression profile of LUAD from The Cancer Genome Atlas (TCGA). R package DiffCorr was implemented for the identification of differential correlations between tumor and adjacent normal tissues. STRING and Cytoscape were used for the construction and visualization of biological networks.
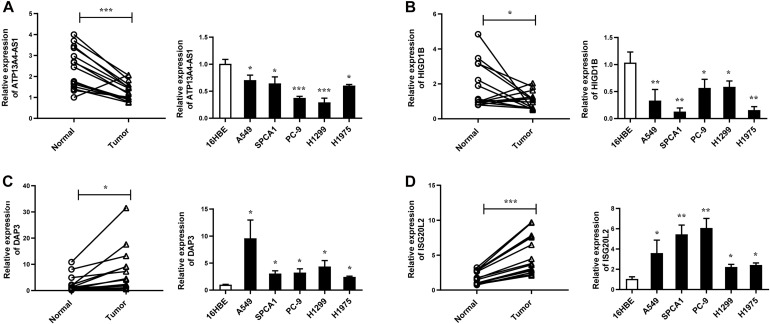
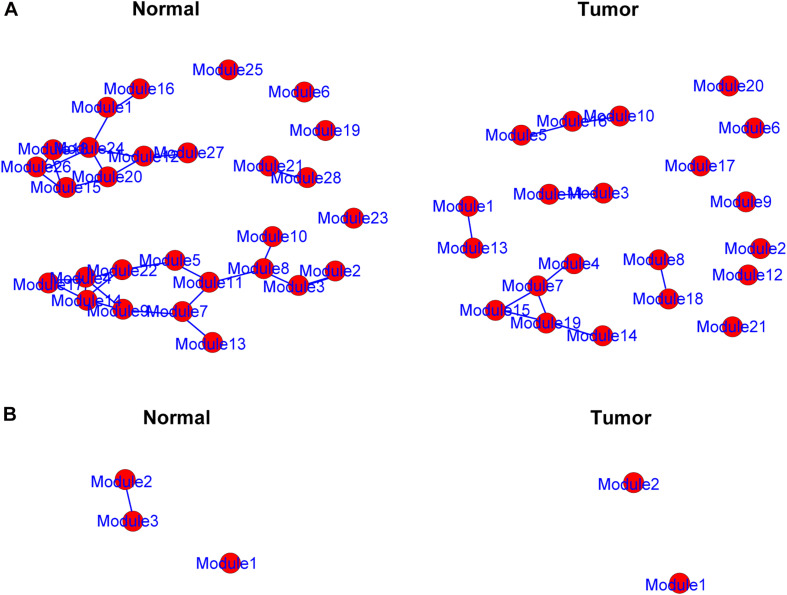
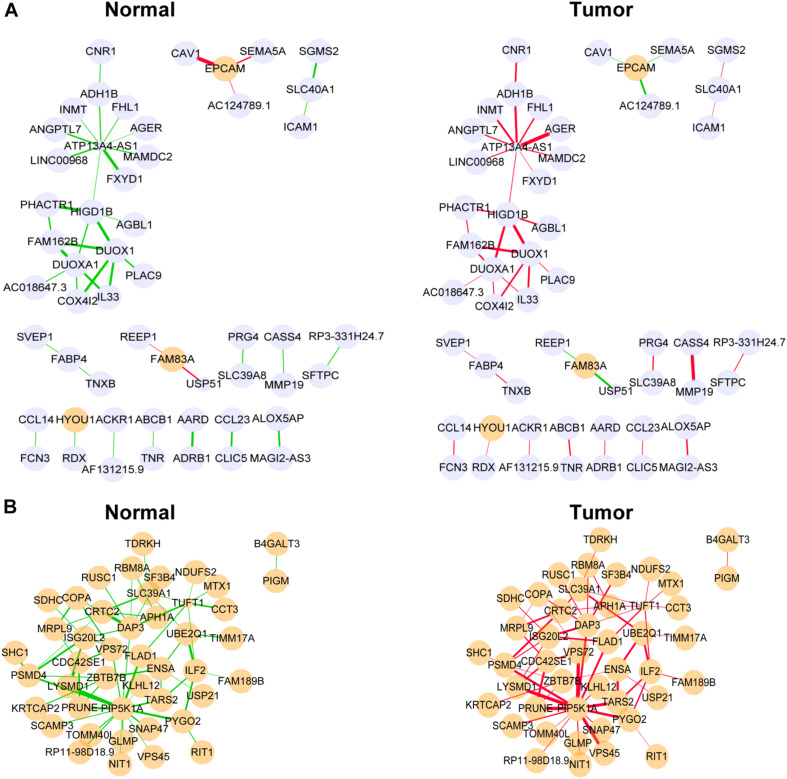
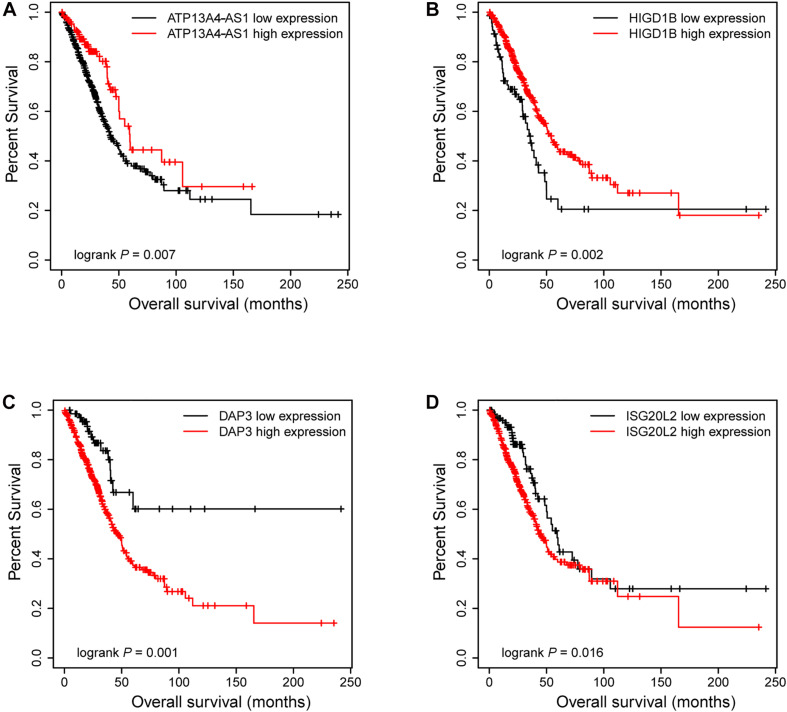
A total of 176 modules were detected in the network, among which yellow and medium orchid modules showed the most significant associations with LUAD. Then genes in these two modules were further chosen to evaluate their differential correlations. Finally, dozens of novel genes with opposite correlations including ATP13A4-AS1, HIGD1B, DAP3, and ISG20L2 were identified. Further biological and survival analyses highlighted their potential values in the diagnosis and treatment of LUAD. Moreover, real-time qPCR confirmed the expression patterns of ATP13A4-AS1, HIGD1B, DAP3, and ISG20L2 in LUAD tissues and cell lines.
Our study provided new insights into the gene regulatory mechanisms during transition from normal to tumor, pioneering a network-based algorithm in the application of tumor etiology.
随着大规模分子谱分析的出现,已鉴定出越来越多有助于精准医学和重塑肺腺癌(LUAD)分类的致癌驱动因素。然而,由于动态突变谱,目前只有少数患者在标准治疗下获得了更好的预后,这就需要扩大易感基因库。越来越多的证据表明,了解基因调控网络及其变化过程有助于识别在致癌过程中起主要调节作用的核心基因。本研究旨在识别正常状态与肿瘤状态之间具有差异相关性的关键基因。
采用加权基因共表达网络分析(WGCNA),利用来自癌症基因组图谱(TCGA)的LUAD表达谱构建基因相互作用网络。使用R包DiffCorr来识别肿瘤组织与相邻正常组织之间的差异相关性。利用STRING和Cytoscape构建和可视化生物网络。
在网络中总共检测到176个模块,其中黄色和中兰花色模块与LUAD的相关性最为显著。然后进一步选择这两个模块中的基因来评估它们的差异相关性。最终,鉴定出了数十个具有相反相关性的新基因,包括ATP13A4-AS1、HIGD1B、DAP3和ISG20L2。进一步的生物学和生存分析突出了它们在LUAD诊断和治疗中的潜在价值。此外,实时定量PCR证实了ATP13A4-AS1、HIGD1B、DAP3和ISG20L2在LUAD组织和细胞系中的表达模式。
我们的研究为从正常到肿瘤转变过程中的基因调控机制提供了新的见解,开创了一种基于网络的算法在肿瘤病因学中的应用。