Zhou Qi, Pang Guofang, Zhang Zhirong, Yuan Huiping, Chen Chen, Zhang Nan, Yang Ze, Sun Liang
The Key Laboratory of Geriatrics, Beijing Institute of Geriatrics, Beijing Hospital, National Center of Gerontology, National Health Commission, Institute of Geriatric Medicine, Chinese Academy of Medical Sciences, Beijing, People's Republic of China.
Guangxi Jiangbin Hospital, Nanning, Guangxi, People's Republic of China.
Diabetes Metab Syndr Obes. 2021 May 17;14:2177-2188. doi: 10.2147/DMSO.S311388. eCollection 2021.
is among the most abundant bacterial species in the human intestine; however, its relationship to metabolic syndrome (MetS)-which is linked to gut dysbiosis-is not known. In this study, we investigated the association between abundance and risk of MetS and its components, as well as dose-response effects and the influence of microbial interactions on the association.
This cross-sectional study included 6896 Chinese participants aged 18 to 97 years from the Guangdong Gut Microbiome Project. MetS was defined according to Joint Committee for Developing Chinese Guidelines on Prevention and Treatment of Dyslipidemia in Adults criteria. The abundance of was assessed by 16S rRNA sequencing. Logistic regression analysis with adjustment for common confounders was performed to evaluate the association between and MetS and its components. Models with restricted cubic splines and interaction terms were used to examine the dose-response association and microbial interactions, respectively.
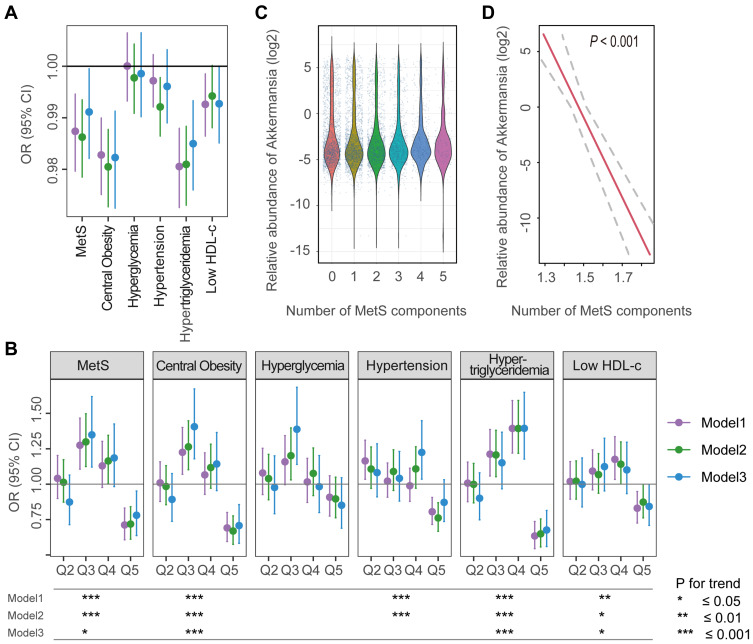
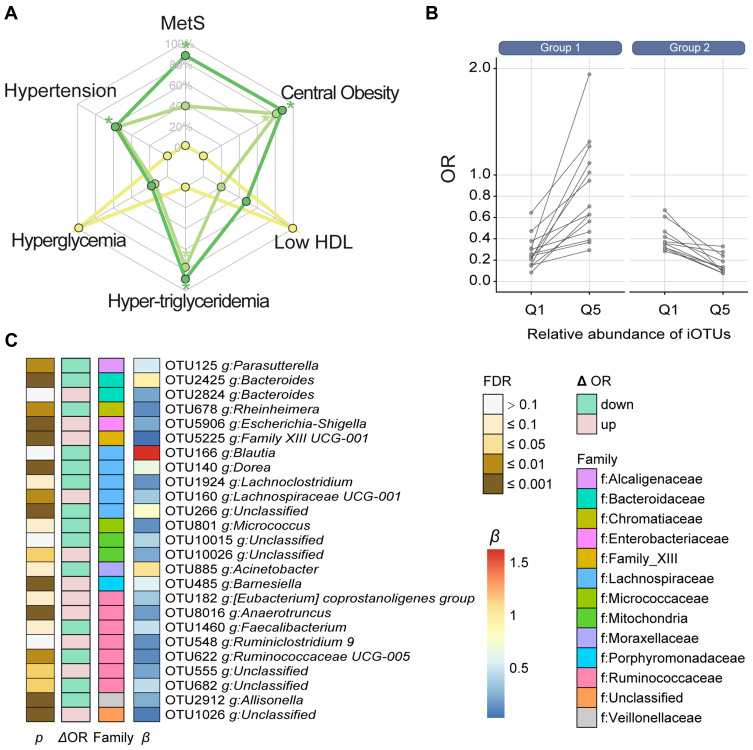
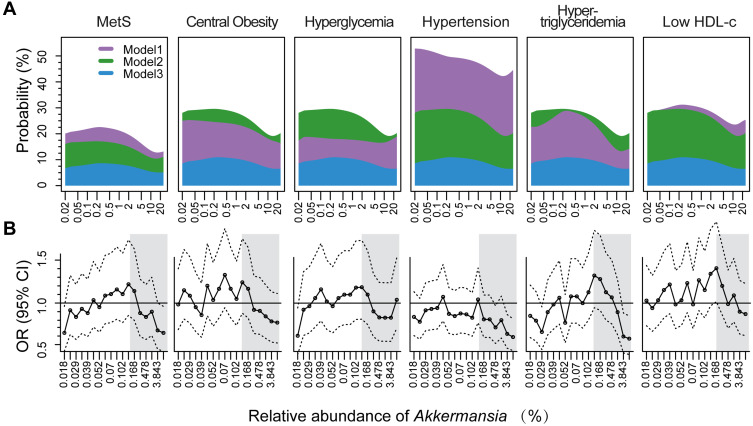
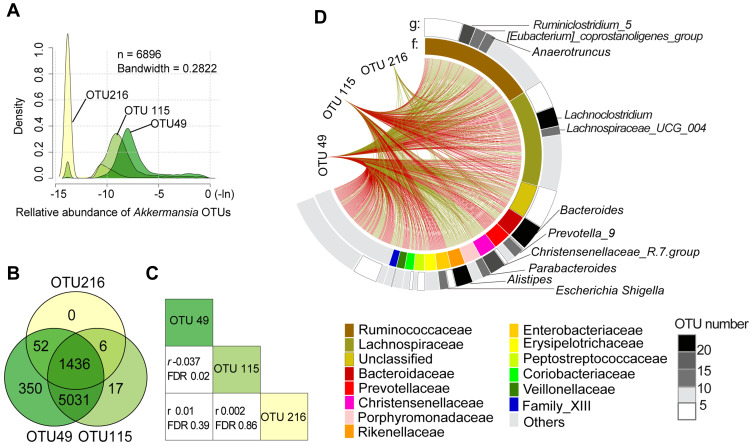
The prevalence of MetS was 20.4%, and the median abundance of was 0.08% (interquartile range: 0.04-0.93%). Increased abundance was associated with decreased risk of MetS ( <0.05), but this effect was not observed until the level was 0.2% of the total gut microbiota abundance (odds ratio=0.96, 95% confidence interval: 0.94-0.98). Of the 5 MetS components, obesity and hypertriglyceridemia showed the strongest association with , followed by reduced high-density lipoprotein cholesterol, hypertension, and hyperglycemia. Microbial interaction analyses showed that Ruminococcaceae and Lachnospiraceae were the predominant bacterial families and were not only correlated with abundance but also influenced the -MetS association.
There is a dose-response association between reduced risk of MetS and increased abundance of . The association between and 5 MetS components is variable and affected by microbial interactions.
是人类肠道中最丰富的细菌物种之一;然而,其与代谢综合征(MetS)(与肠道生态失调相关)的关系尚不清楚。在本研究中,我们调查了其丰度与MetS及其组分风险之间的关联,以及剂量反应效应和微生物相互作用对该关联的影响。
这项横断面研究纳入了来自广东肠道微生物组计划的6896名年龄在18至97岁之间的中国参与者。MetS根据《中国成人血脂异常防治指南联合委员会》标准定义。通过16S rRNA测序评估其丰度。进行了调整常见混杂因素的逻辑回归分析,以评估其与MetS及其组分之间的关联。分别使用具有受限立方样条和交互项的模型来检验剂量反应关联和微生物相互作用。
MetS的患病率为20.4%,其丰度中位数为0.08%(四分位间距:0.04 - 0.93%)。其丰度增加与MetS风险降低相关(P<0.05),但直到其水平达到总肠道微生物群丰度的0.2%时才观察到这种效应(比值比 = 0.96,95%置信区间:0.94 - 0.98)。在5种MetS组分中,肥胖和高甘油三酯血症与它的关联最强,其次是高密度脂蛋白胆固醇降低、高血压和高血糖。微生物相互作用分析表明,瘤胃球菌科和毛螺菌科是主要的细菌科,不仅与它的丰度相关,还影响其与MetS的关联。
MetS风险降低与它的丰度增加之间存在剂量反应关联。它与5种MetS组分之间的关联是可变的,并受微生物相互作用影响。