Xu Yaqin, Dong Yingying, Deng Yunhua, Qi Qianrong, Wu Mi, Liang Hongmei, She Qiuyun, Guo Qing
Department of Dermatology, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, China.
Department of Obstetrics & Gynecology, University of California, Irvine, CA 92697, USA.
Biology (Basel). 2021 May 13;10(5):432. doi: 10.3390/biology10050432.
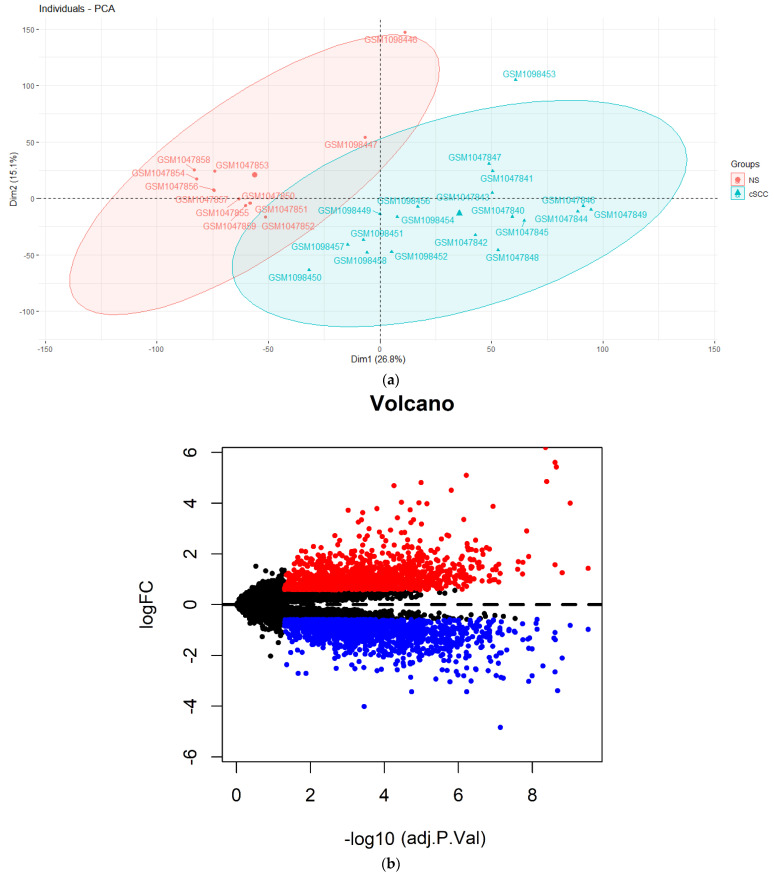
A cutaneous squamous cell carcinoma (cSCC) derived from keratinocytes is the second most common cause of non-melanoma skin cancer. The accumulation of the mutational burden of genes and cellular DNA damage caused by the risk factors (e.g., exposure to ultraviolet radiation) contribute to the aberrant proliferation of keratinocytes and the formation of a cSCC. A cSCC encompasses a spectrum of diseases that range from recursor actinic keratosis (AK) and squamous cell carcinoma (SCC) in situ (SCCIS) to invasive cSCCs and further metastatic SCCs. Emerging evidence has revealed that lncRNAs are involved in the biological process of a cSCC. According to the ceRNA regulatory theory, lncRNAs act as natural miRNA sponges and interact with miRNA response elements, thereby regulating the mRNA expression of their down-stream targets. This study was designed to search for the potential lncRNAs that may become potential therapeutic targets or biomarkers of a cSCC. Considering the spirit of the study to be adequately justified, we collected microarray-based datasets of 19 cSCC tissues and 12 normal skin samples from the GEO database (GSE42677 and GSE45164). After screening the differentially expressed genes via a limma package, we identified 24 differentially expressed lncRNAs (DElncRNAs) and 3221 differentially expressed mRNAs (DEmRNAs). The miRcode, miRTarBase, miRDB and TargetScan databases were used to predict miRNAs that could interact with DElncRNAs and DEmRNAs. A total of 137 miRNA-lncRNA and 221 miRNA-mRNA pairs were retained in the ceRNA network, consisting of 31 miRNAs, 11 DElncRNAs and 155 DEmRNAs. For the functional analysis, the top enriched biological process was enhancer sequence-specific DNA binding in Gene Ontology (GO) terms. The FoxO signaling pathway, autophagy and cellular senescence were the top enrichment terms based on a Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis. The combination of a STRING tool and Cytoscape software (plug-in MCODE) identified five core mRNAs and built a core mRNA-associated ceRNA network. The expression for five identified core mRNAs and their related nine lncRNAs was validated using the external dataset GSE7553. Finally, one lncRNA HLA-F-AS1 and three mRNAs named AGO4, E2F1 and CCND1 were validated with the same expression patterns. We speculate that lncRNA HLA-F-AS1 may sponge miR-17-5p or miR-20b-5p to regulate the expression of CCND1 and E2F1 in the cSCC. The present study may provide potential diagnostic and therapeutic targets for cSCC patients.
源自角质形成细胞的皮肤鳞状细胞癌(cSCC)是非黑素瘤皮肤癌的第二大常见病因。由风险因素(如暴露于紫外线辐射)导致的基因和细胞DNA损伤的突变负担积累,促成了角质形成细胞的异常增殖以及cSCC的形成。cSCC涵盖了一系列疾病,从前驱性光化性角化病(AK)和原位鳞状细胞癌(SCCIS)到侵袭性cSCC以及进一步的转移性SCC。新出现的证据表明lncRNAs参与了cSCC的生物学过程。根据ceRNA调控理论,lncRNAs作为天然的miRNA海绵,与miRNA反应元件相互作用,从而调节其下游靶标的mRNA表达。本研究旨在寻找可能成为cSCC潜在治疗靶点或生物标志物的潜在lncRNAs。考虑到该研究的精神有充分依据,我们从GEO数据库(GSE42677和GSE45164)收集了19个cSCC组织和12个正常皮肤样本的基于微阵列的数据集。通过limma软件包筛选差异表达基因后,我们鉴定出24个差异表达的lncRNAs(DElncRNAs)和3221个差异表达的mRNAs(DEmRNAs)。使用miRcode、miRTarBase、miRDB和TargetScan数据库预测可与DElncRNAs和DEmRNAs相互作用的miRNAs。ceRNA网络中总共保留了137个miRNA-lncRNA对和221个miRNA-mRNA对,由31个miRNAs、11个DElncRNAs和155个DEmRNAs组成。对于功能分析,在基因本体论(GO)术语中最富集的生物学过程是增强子序列特异性DNA结合。基于京都基因与基因组百科全书(KEGG)分析,FoxO信号通路、自噬和细胞衰老为最富集的术语。STRING工具和Cytoscape软件(插件MCODE)的组合鉴定出五个核心mRNAs并构建了一个核心mRNA相关的ceRNA网络。使用外部数据集GSE7553验证了五个鉴定出的核心mRNAs及其相关的九个lncRNAs的表达。最后,一个lncRNA HLA-F-AS1和三个名为AGO4、E2F1和CCND1的mRNAs被验证具有相同的表达模式。我们推测lncRNA HLA-F-AS1可能通过吸附miR-17-5p或miR-20b-5p来调节cSCC中CCND1和EIF1的表达。本研究可能为cSCC患者提供潜在的诊断和治疗靶点。