Department of Physiology, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104.
Aligning Science Across Parkinson's Collaborative Research Network, Chevy Chase, MD 20815.
Proc Natl Acad Sci U S A. 2021 Jun 15;118(24). doi: 10.1073/pnas.2025053118.
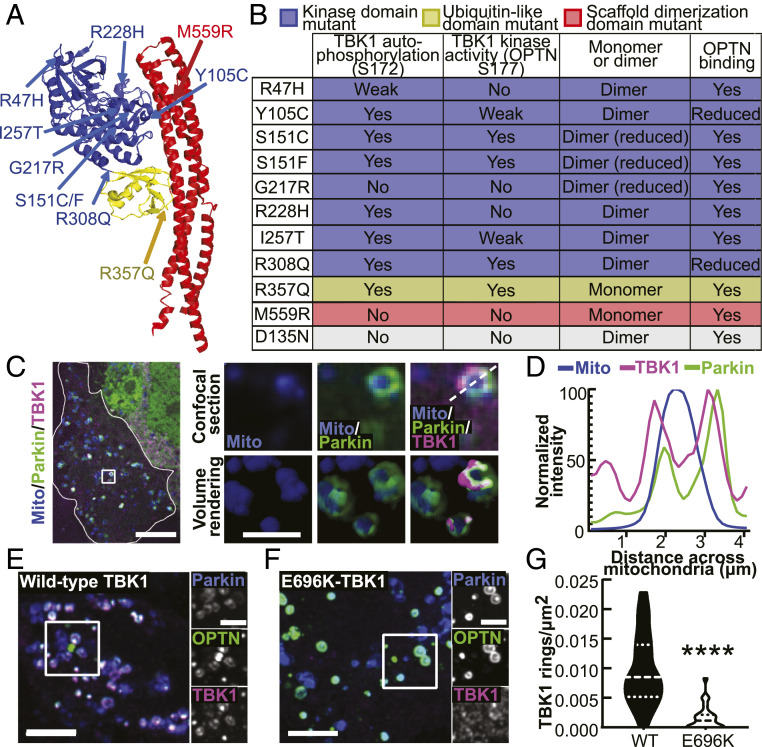
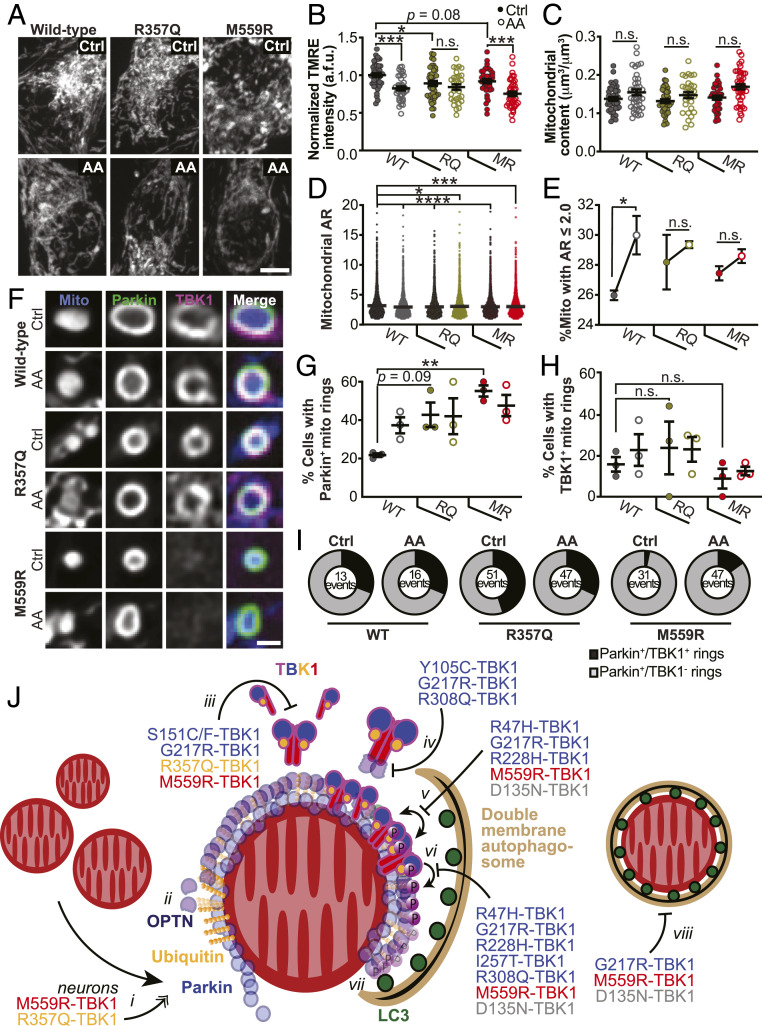
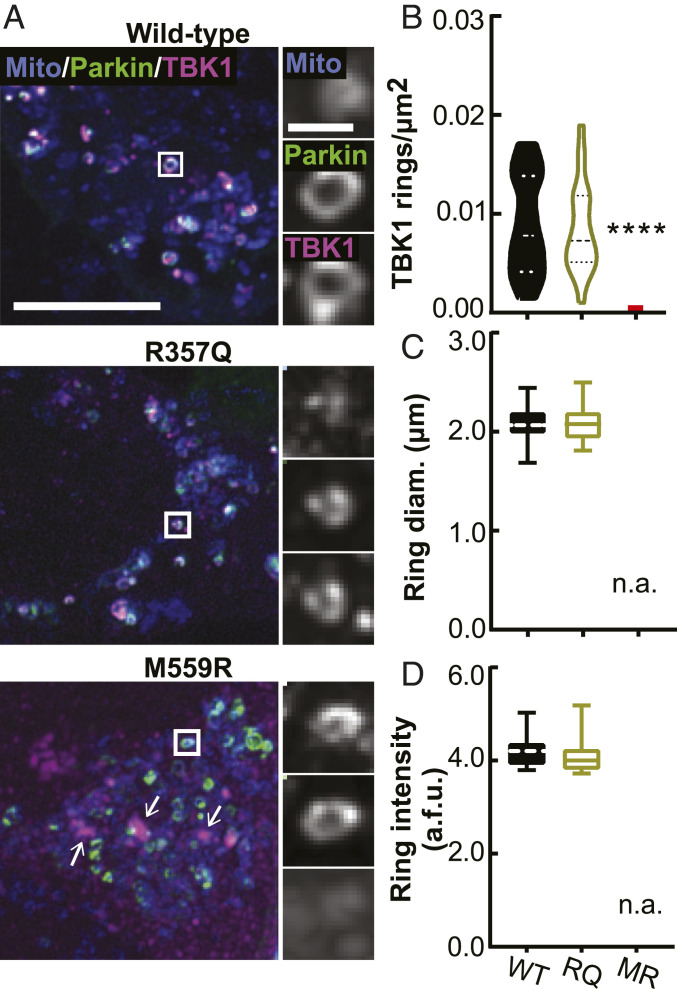
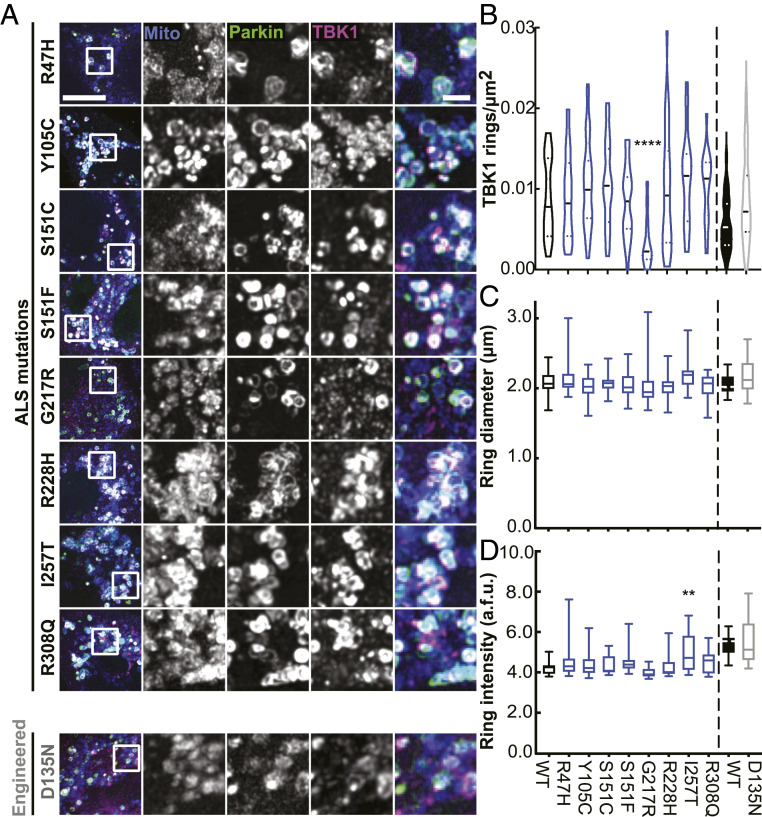
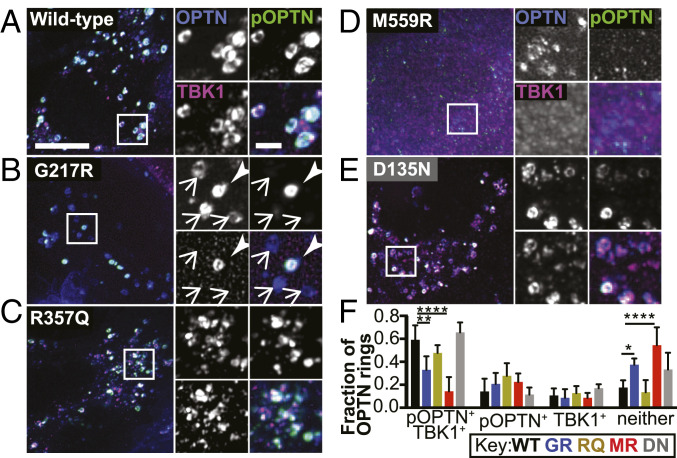
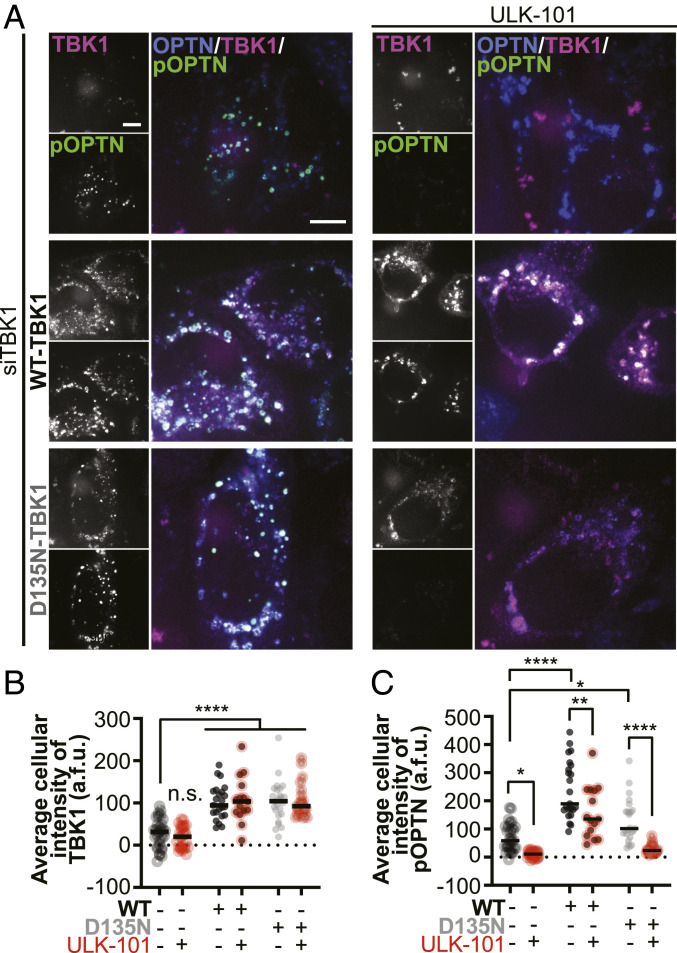
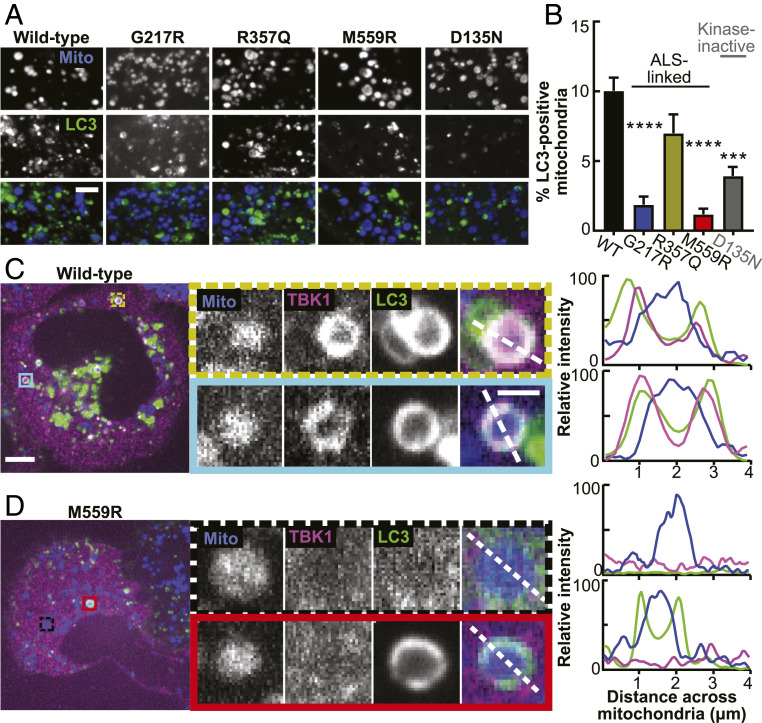
TANK-binding kinase 1 (TBK1) is a multifunctional kinase with an essential role in mitophagy, the selective clearance of damaged mitochondria. More than 90 distinct mutations in TBK1 are linked to amyotrophic lateral sclerosis (ALS) and fronto-temporal dementia, including missense mutations that disrupt the abilities of TBK1 to dimerize, associate with the mitophagy receptor optineurin (OPTN), autoactivate, or catalyze phosphorylation. We investigated how ALS-associated mutations in TBK1 affect Parkin-dependent mitophagy using imaging to dissect the molecular mechanisms involved in clearing damaged mitochondria. Some mutations cause severe dysregulation of the pathway, while others induce limited disruption. Mutations that abolish either TBK1 dimerization or kinase activity were insufficient to fully inhibit mitophagy, while mutations that reduced both dimerization and kinase activity were more disruptive. Ultimately, both TBK1 recruitment and OPTN phosphorylation at S177 are necessary for engulfment of damaged mitochondra by autophagosomal membranes. Surprisingly, we find that ULK1 activity contributes to the phosphorylation of OPTN in the presence of either wild-type or kinase-inactive TBK1. In primary neurons, TBK1 mutants induce mitochondrial stress under basal conditions; network stress is exacerbated with further mitochondrial insult. Our study further refines the model for TBK1 function in mitophagy, demonstrating that some ALS-linked mutations likely contribute to disease pathogenesis by inducing mitochondrial stress or inhibiting mitophagic flux. Other TBK1 mutations exhibited much less impact on mitophagy in our assays, suggesting that cell-type-specific effects, cumulative damage, or alternative TBK1-dependent pathways such as innate immunity and inflammation also factor into the development of ALS in affected individuals.
TANK 结合激酶 1(TBK1)是一种多功能激酶,在自噬中起着重要作用,自噬是一种选择性清除受损线粒体的过程。超过 90 种不同的 TBK1 突变与肌萎缩侧索硬化症(ALS)和额颞叶痴呆有关,包括导致 TBK1 二聚化、与自噬受体视神经萎缩症(OPTN)结合、自动激活或催化磷酸化能力受损的错义突变。我们研究了 ALS 相关的 TBK1 突变如何影响 Parkin 依赖性的自噬,使用成像技术来剖析参与清除受损线粒体的分子机制。一些突变导致途径严重失调,而另一些突变则导致有限的破坏。消除 TBK1 二聚化或激酶活性的突变不足以完全抑制自噬,而降低二聚化和激酶活性的突变则更具破坏性。最终,TBK1 募集和 OPTN 在 S177 处的磷酸化对于自噬体膜吞噬受损线粒体都是必要的。令人惊讶的是,我们发现,即使存在野生型或激酶失活的 TBK1,ULK1 活性也有助于 OPTN 的磷酸化。在原代神经元中,TBK1 突变体在基础条件下诱导线粒体应激;进一步的线粒体损伤会加剧网络应激。我们的研究进一步完善了 TBK1 在自噬中的作用模型,表明一些与 ALS 相关的突变可能通过诱导线粒体应激或抑制自噬通量来导致疾病的发生。在我们的实验中,其他 TBK1 突变对自噬的影响要小得多,这表明细胞类型特异性效应、累积损伤或替代的 TBK1 依赖性途径,如先天免疫和炎症,也会影响受影响个体中 ALS 的发展。