Department of Life Sciences, Imperial College London, London, UK.
Department of Cell Biology, Embryology and Genetics, Federal University of Santa Catarina (UFSC), Florianópolis, Brazil.
BMC Genomics. 2021 Jun 8;22(1):422. doi: 10.1186/s12864-021-07722-y.
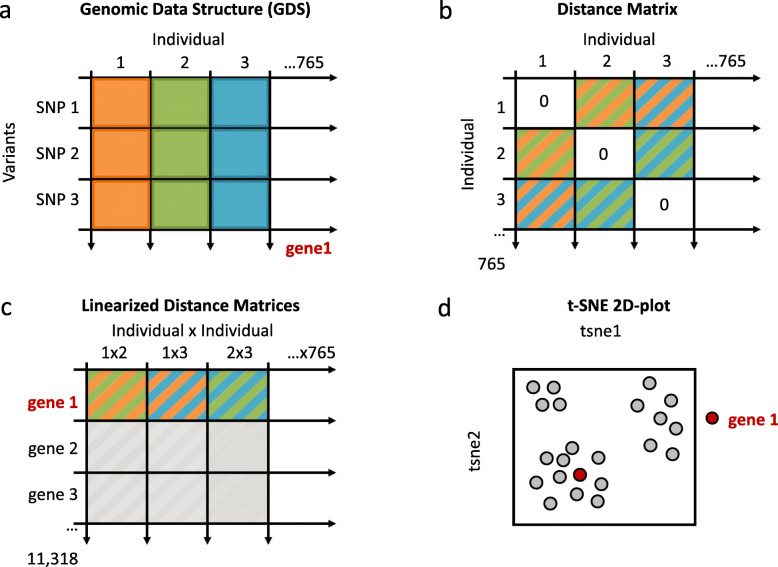
Whole genome re-sequencing provides powerful data for population genomic studies, allowing robust inferences of population structure, gene flow and evolutionary history. For the major malaria vector in Africa, Anopheles gambiae, other genetic aspects such as selection and adaptation are also important. In the present study, we explore population genetic variation from genome-wide sequencing of 765 An. gambiae and An. coluzzii specimens collected from across Africa. We used t-SNE, a recently popularized dimensionality reduction method, to create a 2D-map of An. gambiae and An. coluzzii genes that reflect their population structure similarities.
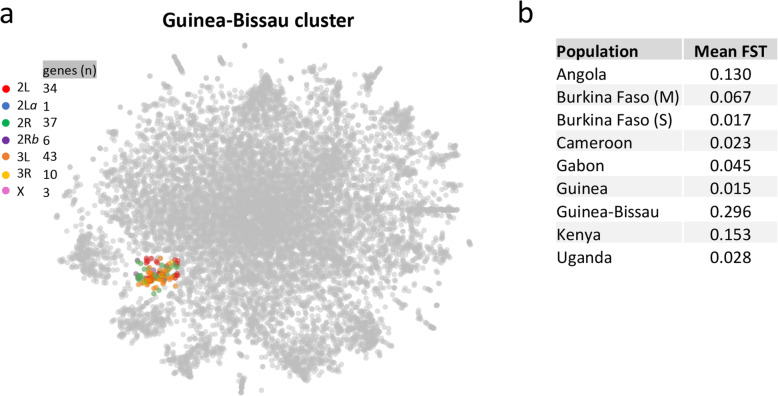
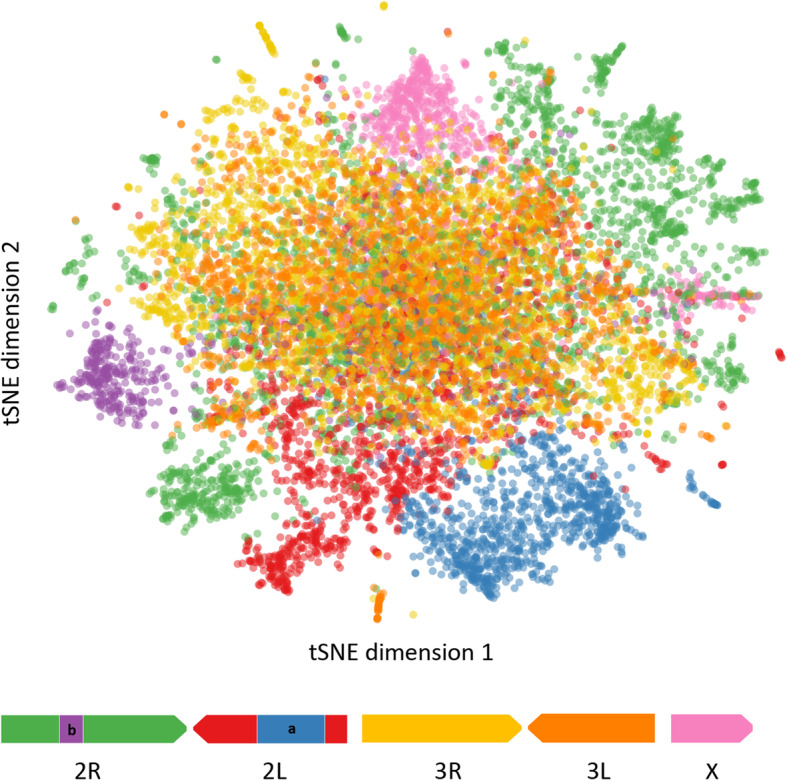
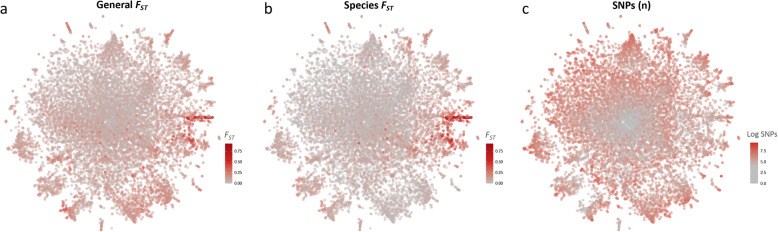
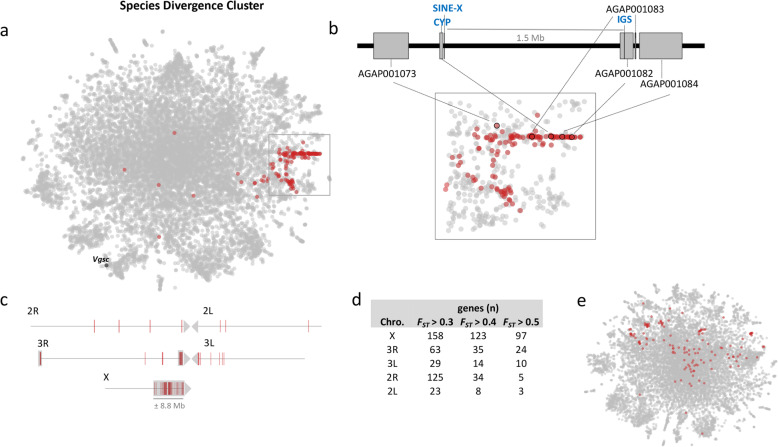
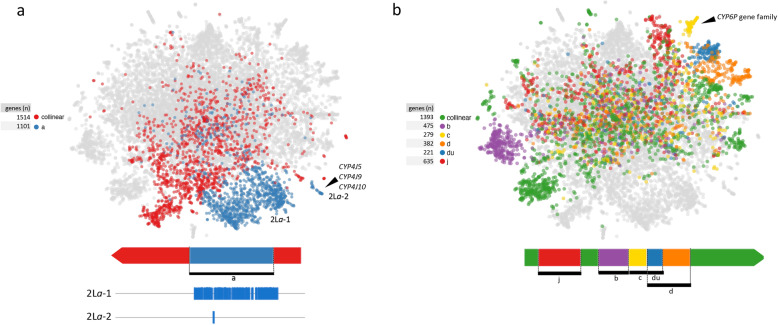
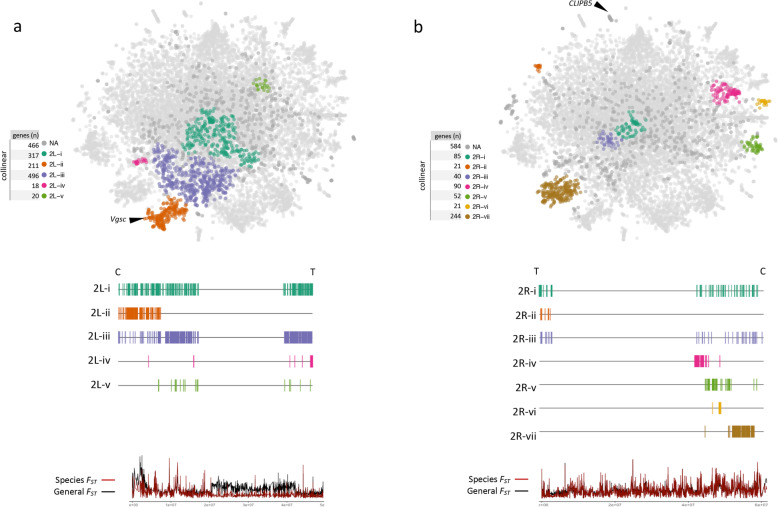
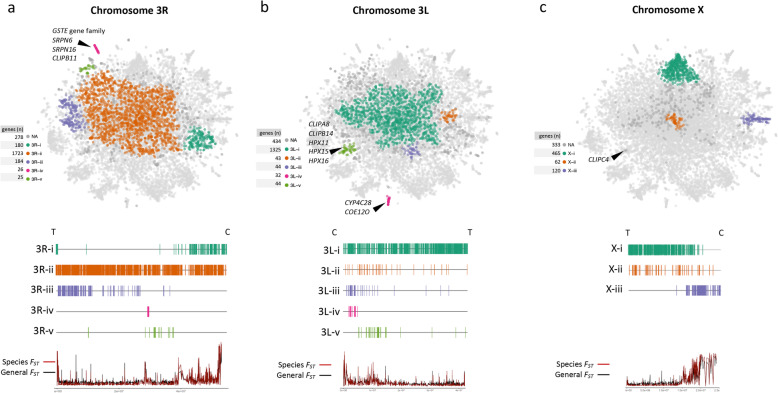
The map allows intuitive navigation among genes distributed throughout the so-called "mainland" and numerous surrounding "island-like" gene clusters. These gene clusters of various sizes correspond predominantly to low recombination genomic regions such as inversions and centromeres, and also to recent selective sweeps. Because this mosquito species complex has been studied extensively, we were able to support our interpretations with previously published findings. Several novel observations and hypotheses are also made, including selective sweeps and a multi-locus selection event in Guinea-Bissau, a known intense hybridization zone between An. gambiae and An. coluzzii.
Our results present a rich dataset that could be utilized in functional investigations aiming to shed light onto An. gambiae s.l genome evolution and eventual speciation. In addition, the methodology presented here can be used to further characterize other species not so well studied as An. gambiae, shortening the time required to progress from field sampling to the identification of genes and genomic regions under unique evolutionary processes.
全基因组重测序为群体基因组研究提供了强大的数据支持,使人们能够对群体结构、基因流和进化历史进行强有力的推断。对于非洲主要的疟疾媒介——冈比亚按蚊(Anopheles gambiae)来说,其他遗传方面,如选择和适应,也很重要。在本研究中,我们探索了来自非洲各地采集的 765 只冈比亚按蚊(An. gambiae)和库蚊(An. coluzzii)标本的全基因组测序中的群体遗传变异。我们使用了 t-SNE,这是一种最近流行的降维方法,创建了一个反映冈比亚按蚊和库蚊基因群体结构相似性的二维图谱。
该图谱允许在分布在所谓“大陆”和许多周围“岛屿状”基因簇中的基因之间进行直观导航。这些大小不一的基因簇主要对应于低重组基因组区域,如倒位和着丝粒,也对应于最近的选择清除。由于这个蚊种复合体已经被广泛研究,我们能够用以前发表的发现来支持我们的解释。我们还提出了一些新的观察和假设,包括在几内亚比绍的选择清除和多基因选择事件,几内亚比绍是冈比亚按蚊和库蚊之间已知的强烈杂交区。
我们的研究结果提供了一个丰富的数据集,可用于功能研究,旨在阐明冈比亚按蚊 s.l. 基因组的进化和最终的物种形成。此外,这里提出的方法可用于进一步描述其他研究不如冈比亚按蚊充分的物种,从而缩短从实地采样到识别独特进化过程中的基因和基因组区域的时间。