Department of Medicine, Division of Hematology/Oncology, O'Neal Comprehensive Cancer Center, University of Alabama at Birmingham, Birmingham, AL, USA.
Department of Biochemistry and Molecular Genetics, School of Medicine, University of Colorado Anschutz Medical Campus, Aurora, CO, USA.
Leukemia. 2021 Dec;35(12):3371-3382. doi: 10.1038/s41375-021-01315-0. Epub 2021 Jun 12.
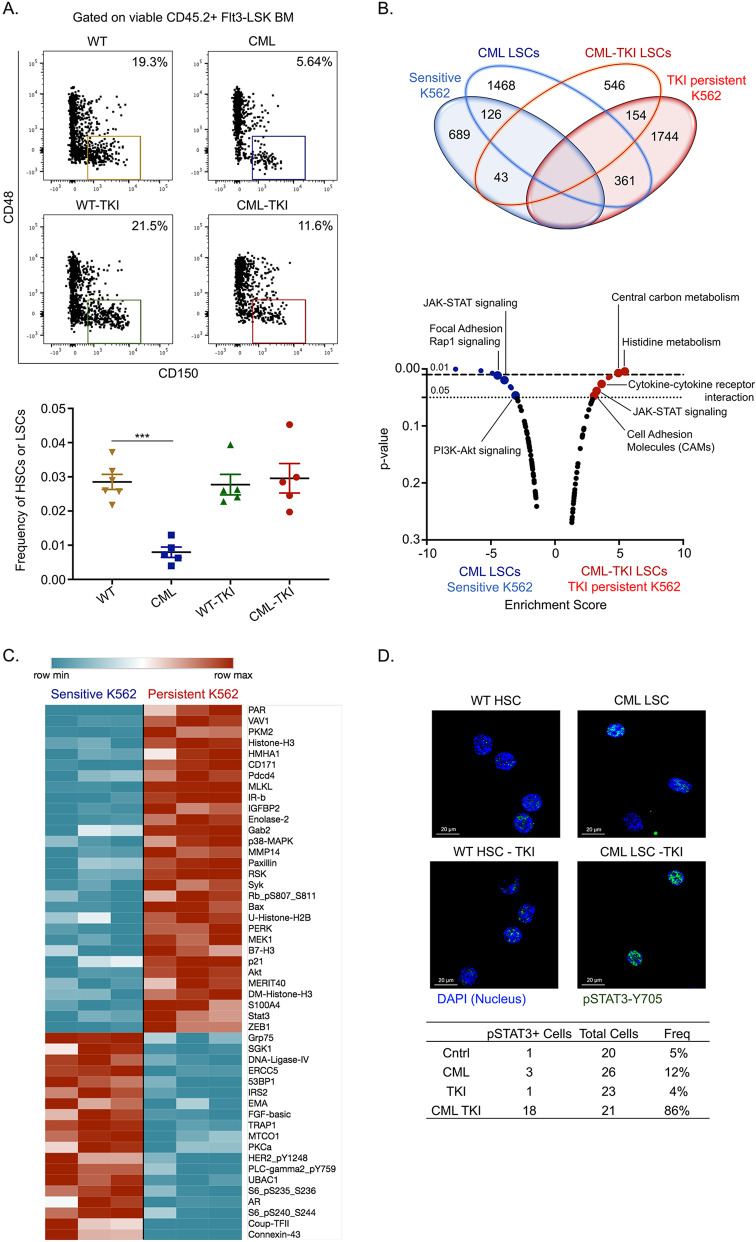
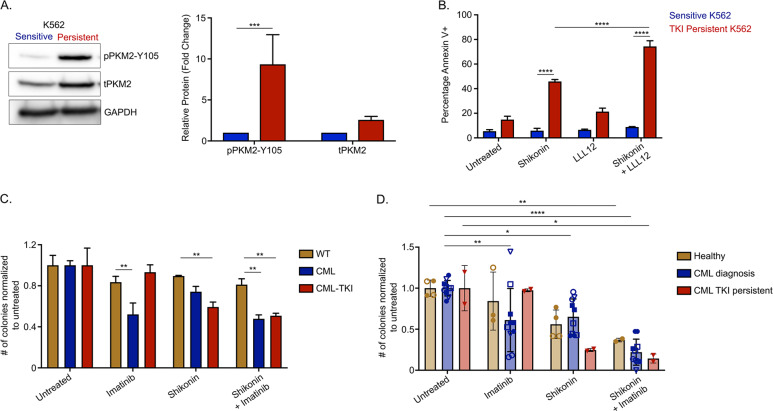
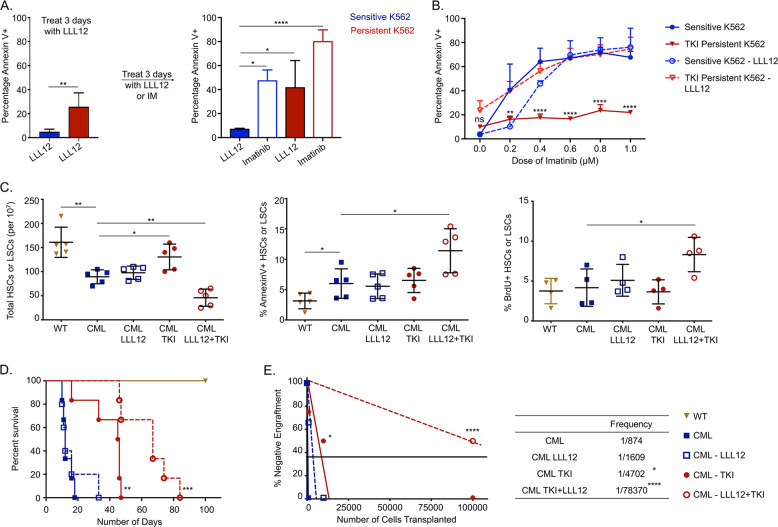
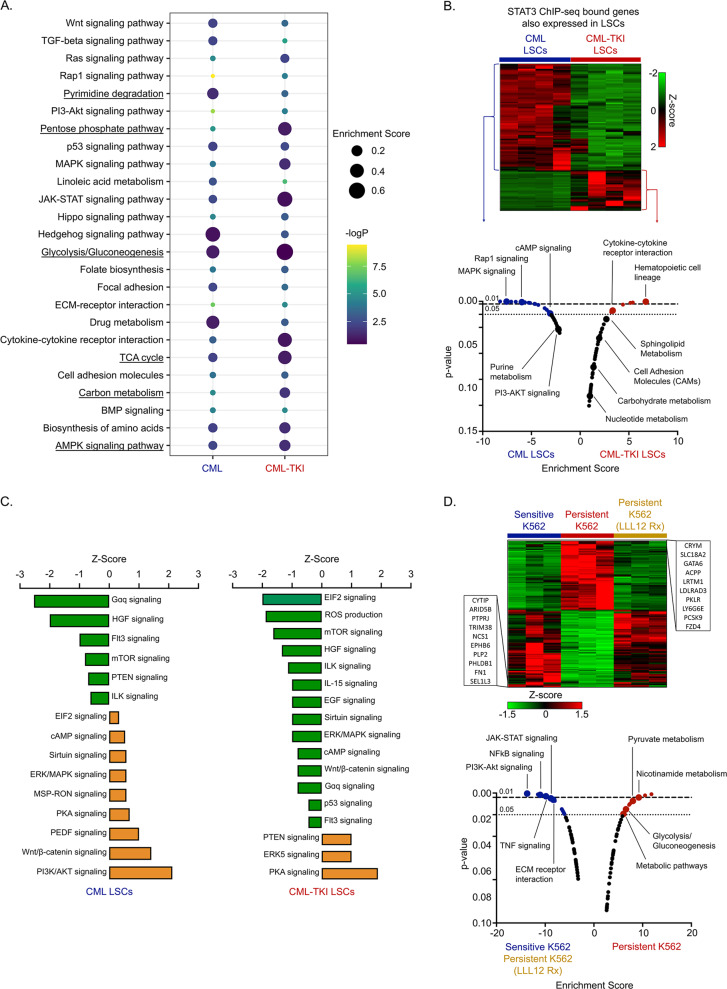
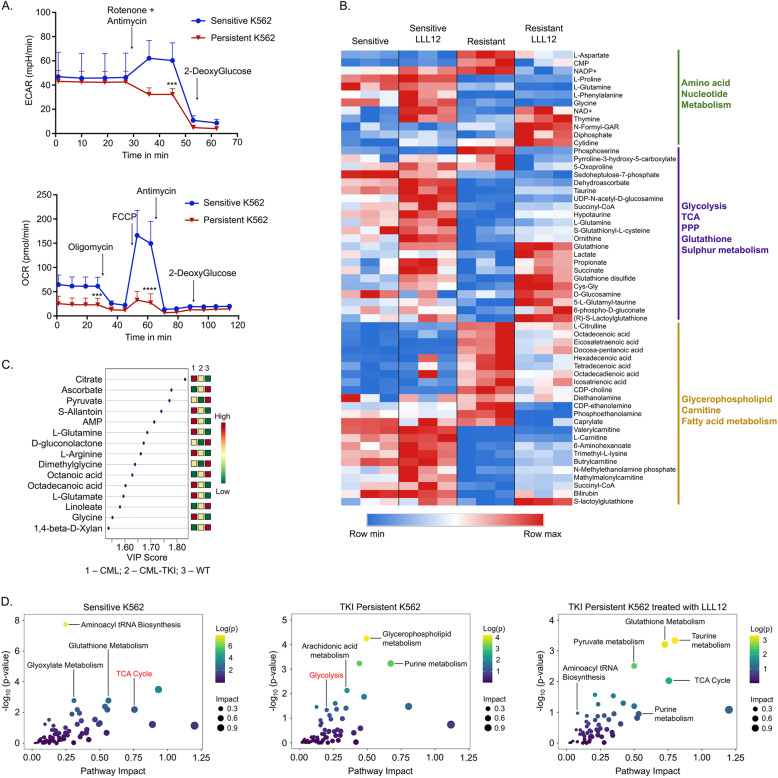
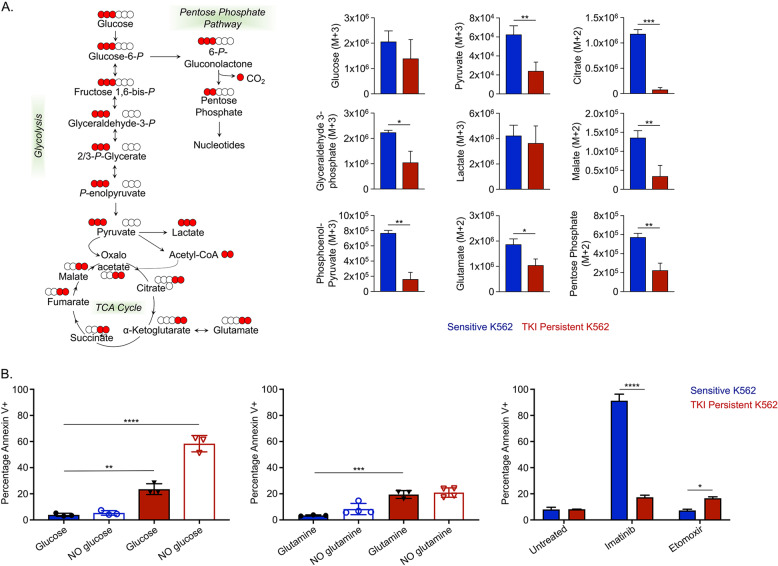
Leukemic stem cells (LSCs) can acquire non-mutational resistance following drug treatment leading to therapeutic failure and relapse. However, oncogene-independent mechanisms of drug persistence in LSCs are incompletely understood, which is the primary focus of this study. We integrated proteomics, transcriptomics, and metabolomics to determine the contribution of STAT3 in promoting metabolic changes in tyrosine kinase inhibitor (TKI) persistent chronic myeloid leukemia (CML) cells. Proteomic and transcriptional differences in TKI persistent CML cells revealed BCR-ABL-independent STAT3 activation in these cells. While knockout of STAT3 inhibited the CML cells from developing drug-persistence, inhibition of STAT3 using a small molecule inhibitor sensitized the persistent CML cells to TKI treatment. Interestingly, given the role of phosphorylated STAT3 as a transcription factor, it localized uniquely to genes regulating metabolic pathways in the TKI-persistent CML stem and progenitor cells. Subsequently, we observed that STAT3 dysregulated mitochondrial metabolism forcing the TKI-persistent CML cells to depend on glycolysis, unlike TKI-sensitive CML cells, which are more reliant on oxidative phosphorylation. Finally, targeting pyruvate kinase M2, a rate-limiting glycolytic enzyme, specifically eradicated the TKI-persistent CML cells. By exploring the role of STAT3 in altering metabolism, we provide critical insight into identifying potential therapeutic targets for eliminating TKI-persistent LSCs.
白血病干细胞(LSCs)在药物治疗后可以获得非突变性耐药性,从而导致治疗失败和复发。然而,LSCs 中与癌基因无关的药物持续存在的机制尚未完全了解,这是本研究的主要重点。我们整合了蛋白质组学、转录组学和代谢组学,以确定 STAT3 在促进酪氨酸激酶抑制剂(TKI)持续慢性髓性白血病(CML)细胞代谢变化中的作用。TKI 持续存在的 CML 细胞中的蛋白质组学和转录组学差异揭示了这些细胞中 BCR-ABL 独立的 STAT3 激活。虽然 STAT3 的敲除抑制了 CML 细胞产生药物耐药性,但使用小分子抑制剂抑制 STAT3 使持续存在的 CML 细胞对 TKI 治疗敏感。有趣的是,鉴于磷酸化 STAT3 作为转录因子的作用,它仅定位于调节 TKI 持续存在的 CML 干细胞和祖细胞中代谢途径的基因。随后,我们观察到 STAT3 失调线粒体代谢,迫使 TKI 持续存在的 CML 细胞依赖于糖酵解,而不像 TKI 敏感的 CML 细胞,后者更依赖于氧化磷酸化。最后,靶向丙酮酸激酶 M2,一种限速糖酵解酶,特异性根除了 TKI 持续存在的 CML 细胞。通过探索 STAT3 在改变代谢中的作用,我们深入了解确定消除 TKI 持续存在的 LSCs 的潜在治疗靶点。