Diabetes Research Program, Department of Medicine, New York University Grossman School of Medicine, New York, NY, 10016, USA.
Department of Human Physiology and Pathophysiology, School of Medicine, University of Warmia and Mazury, Olsztyn, Poland.
J Neuroinflammation. 2021 Jun 15;18(1):139. doi: 10.1186/s12974-021-02191-2.
Burgeoning evidence highlights seminal roles for microglia in the pathogenesis of neurodegenerative diseases including amyotrophic lateral sclerosis (ALS). The receptor for advanced glycation end products (RAGE) binds ligands relevant to ALS that accumulate in the diseased spinal cord and RAGE has been previously implicated in the progression of ALS pathology.
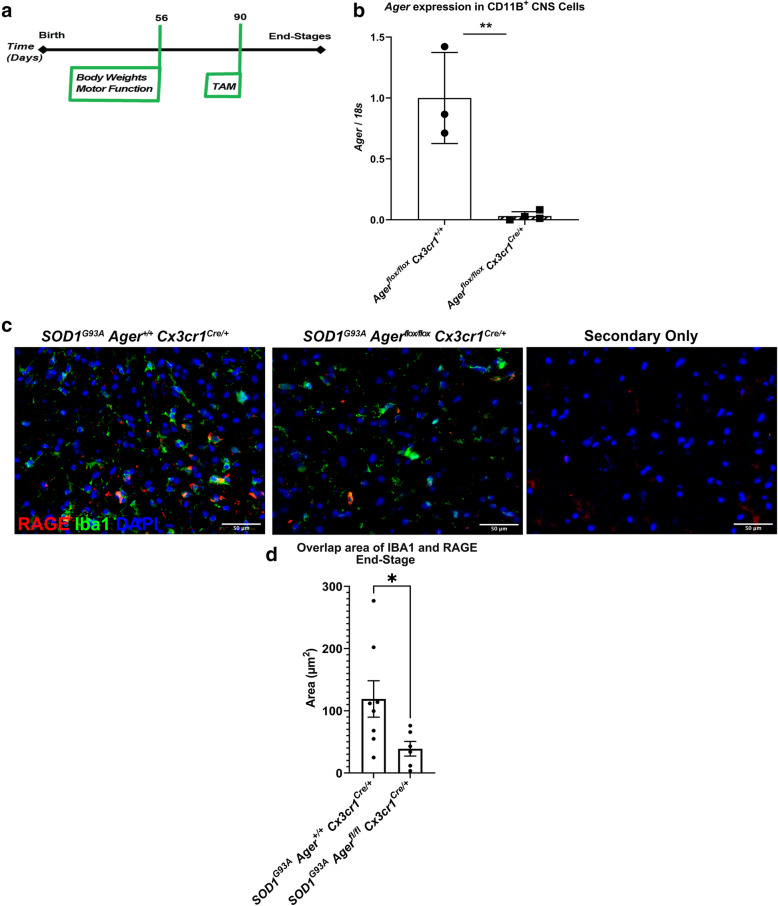
We generated a novel mouse model to temporally delete Ager from microglia in the murine SOD1 model of ALS. Microglia Ager deficient SOD1 mice and controls were examined for changes in survival, motor function, gliosis, motor neuron numbers, and transcriptomic analyses of lumbar spinal cord. Furthermore, we examined bulk-RNA-sequencing transcriptomic analyses of human ALS cervical spinal cord.
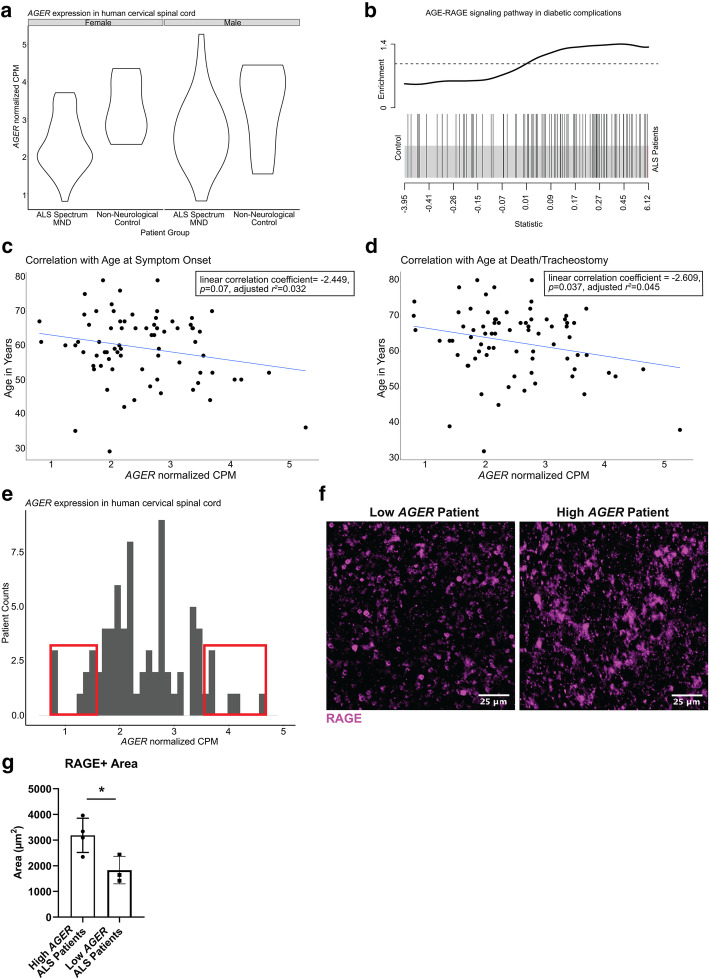
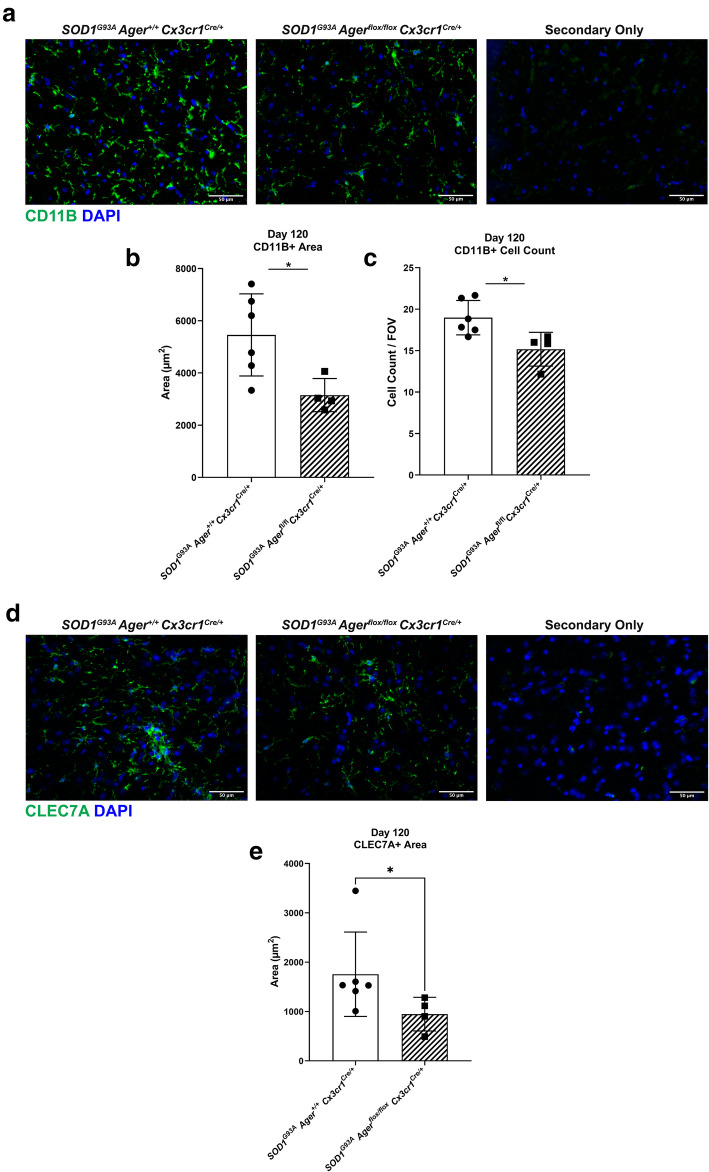
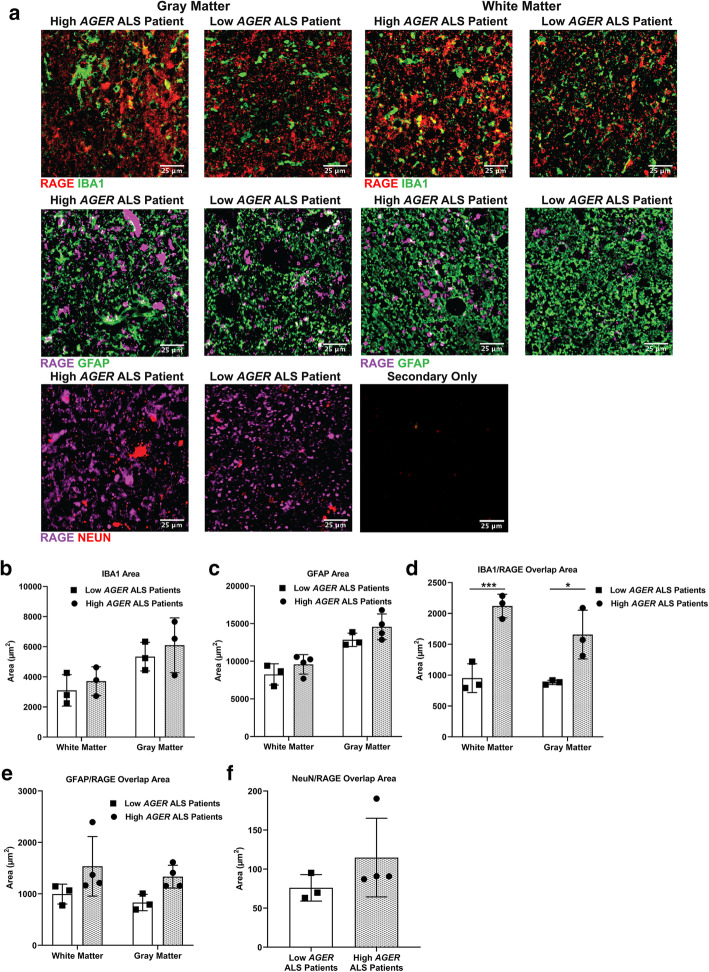
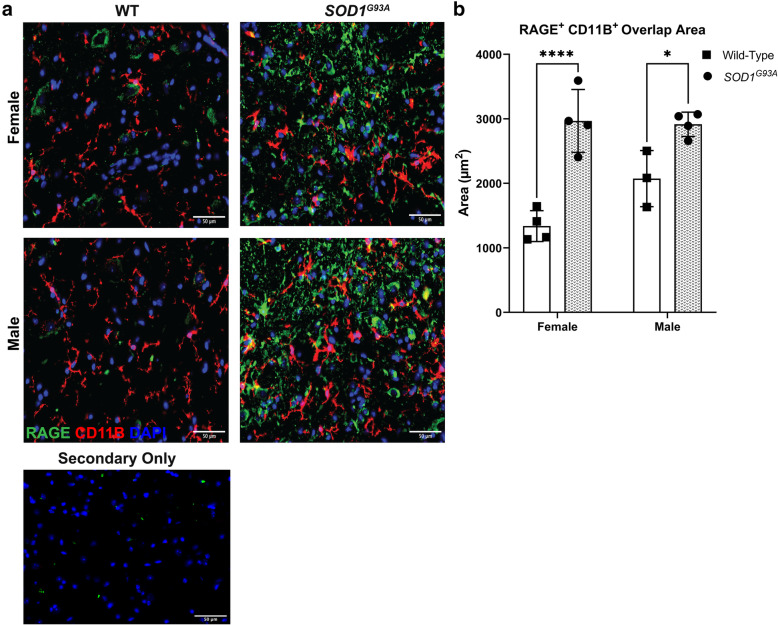
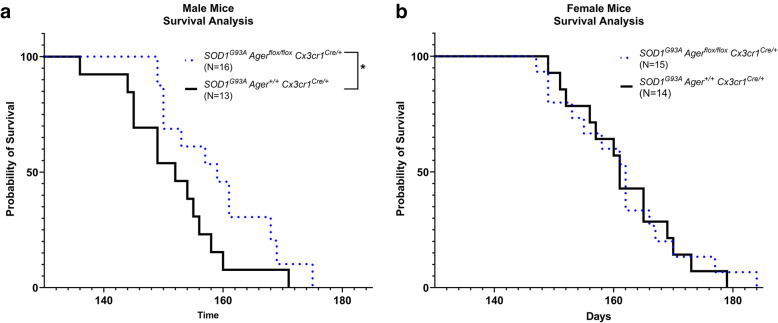
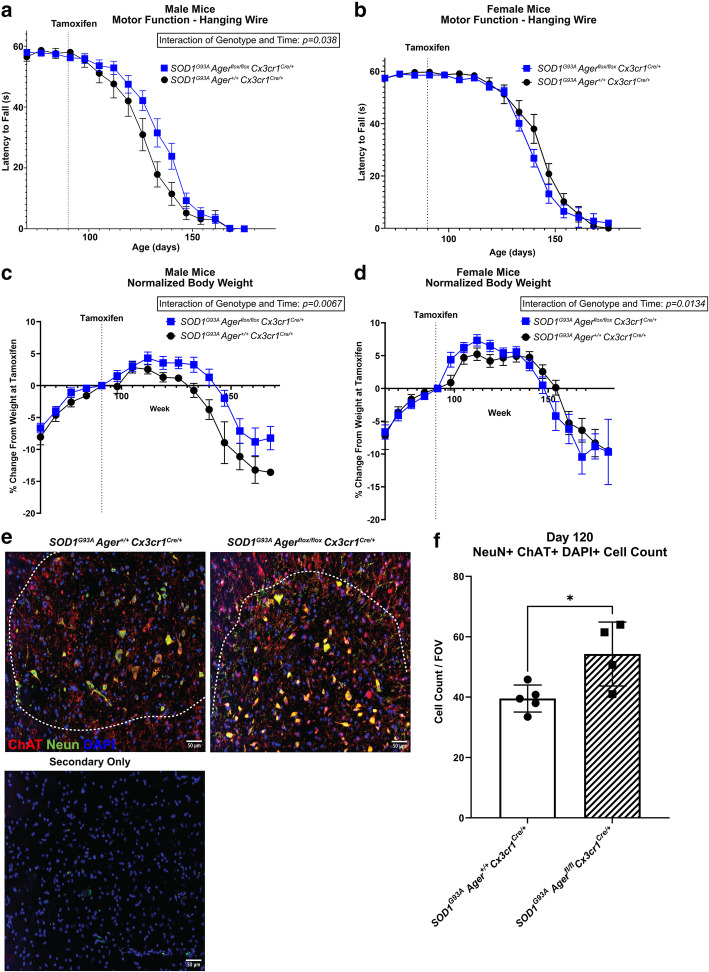
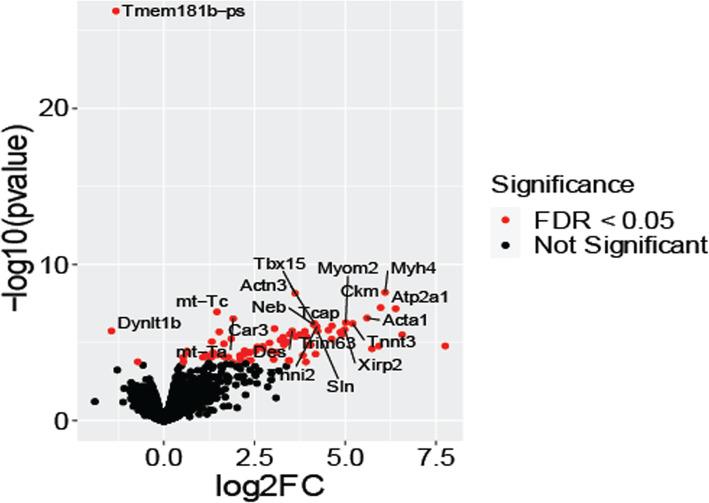
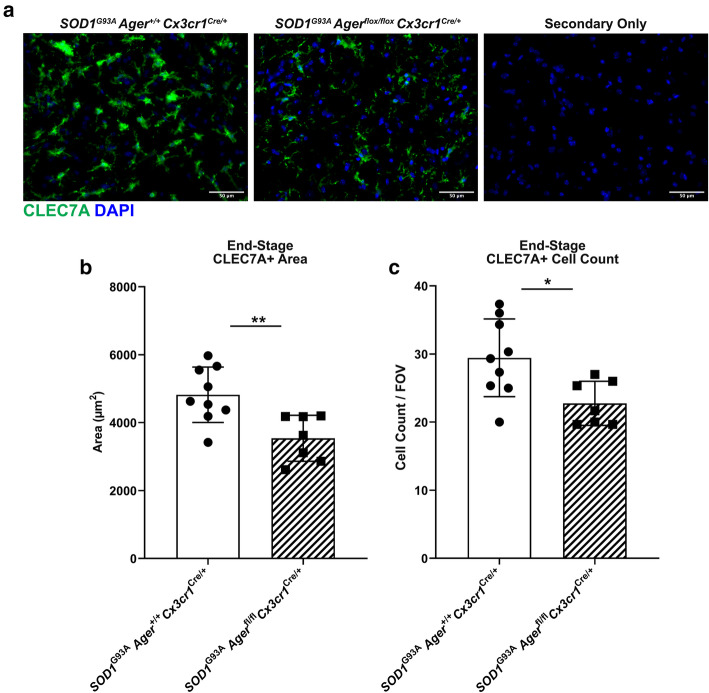
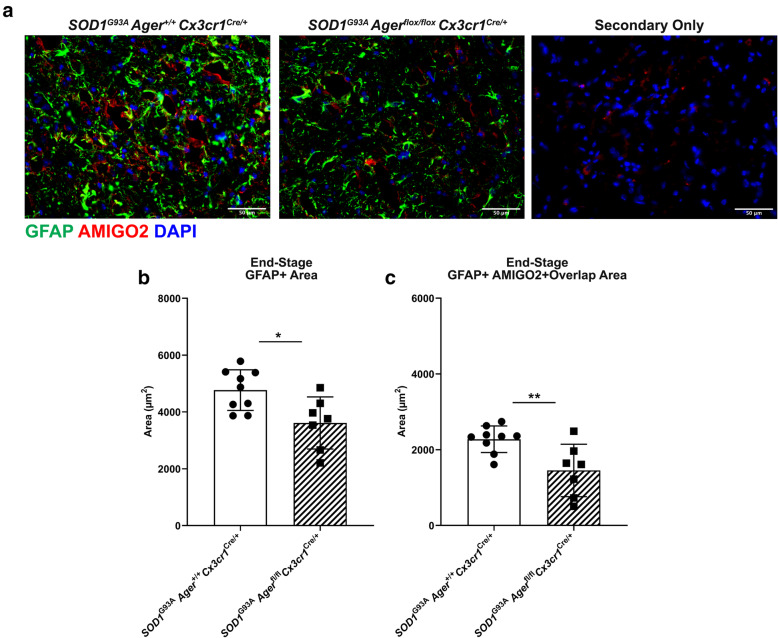
Transcriptomic analysis of human cervical spinal cord reveals a range of AGER expression in ALS patients, which was negatively correlated with age at disease onset and death or tracheostomy. The degree of AGER expression related to differential expression of pathways involved in extracellular matrix, lipid metabolism, and intercellular communication. Microglia display increased RAGE immunoreactivity in the spinal cords of high AGER expressing patients and in the SOD1 murine model of ALS vs. respective controls. We demonstrate that microglia Ager deletion at the age of symptomatic onset, day 90, in SOD1 mice extends survival in male but not female mice. Critically, many of the pathways identified in human ALS patients that accompanied increased AGER expression were significantly ameliorated by microglia Ager deletion in male SOD1 mice.
Our results indicate that microglia RAGE disrupts communications with cell types including astrocytes and neurons, intercellular communication pathways that divert microglia from a homeostatic to an inflammatory and tissue-injurious program. In totality, microglia RAGE contributes to the progression of SOD1 murine pathology in male mice and may be relevant in human disease.
越来越多的证据表明小胶质细胞在神经退行性疾病(包括肌萎缩侧索硬化症,ALS)的发病机制中具有重要作用。晚期糖基化终产物受体(RAGE)与 ALS 相关的配体结合,这些配体在患病的脊髓中积累,而 RAGE 先前与 ALS 病理学的进展有关。
我们构建了一种新的小鼠模型,用于在 SOD1 型 ALS 小鼠模型中,在时间上从小胶质细胞中敲除 Ager。研究了小胶质细胞 Ager 缺陷的 SOD1 小鼠和对照组在生存、运动功能、神经胶质增生、运动神经元数量以及腰椎脊髓的转录组分析方面的变化。此外,我们还研究了人类 ALS 颈段脊髓的大量 RNA 测序转录组分析。
人类颈段脊髓的转录组分析显示,ALS 患者的 AGER 表达存在多种差异,与疾病发病年龄和死亡或气管切开呈负相关。AGER 表达的程度与涉及细胞外基质、脂质代谢和细胞间通讯的途径的差异表达相关。在高 AGER 表达的患者的脊髓和 SOD1 型 ALS 小鼠模型中,小胶质细胞的 RAGE 免疫反应性增加。我们证明,在 SOD1 小鼠中,在症状发作时(第 90 天)敲除小胶质细胞 Ager,可延长雄性而非雌性小鼠的生存期。重要的是,在雄性 SOD1 小鼠中敲除小胶质细胞 Ager,可显著改善人类 ALS 患者中伴随 AGER 表达增加的许多通路。
我们的结果表明,小胶质细胞 RAGE 破坏了与包括星形胶质细胞和神经元在内的细胞类型之间的通讯,改变了小胶质细胞的细胞间通讯途径,使小胶质细胞从稳态转变为炎症和组织损伤的程序。总之,小胶质细胞 RAGE 促进了雄性 SOD1 小鼠的病理学进展,可能与人类疾病有关。