Institute for Quantitative and Computational Biosciences (QCBio), University of California, Los Angeles, California, United States of America.
Department of Microbiology, Immunology, and Molecular Genetics, University of California, Los Angeles, California, United States of America.
PLoS Comput Biol. 2021 Jun 24;17(6):e1009095. doi: 10.1371/journal.pcbi.1009095. eCollection 2021 Jun.
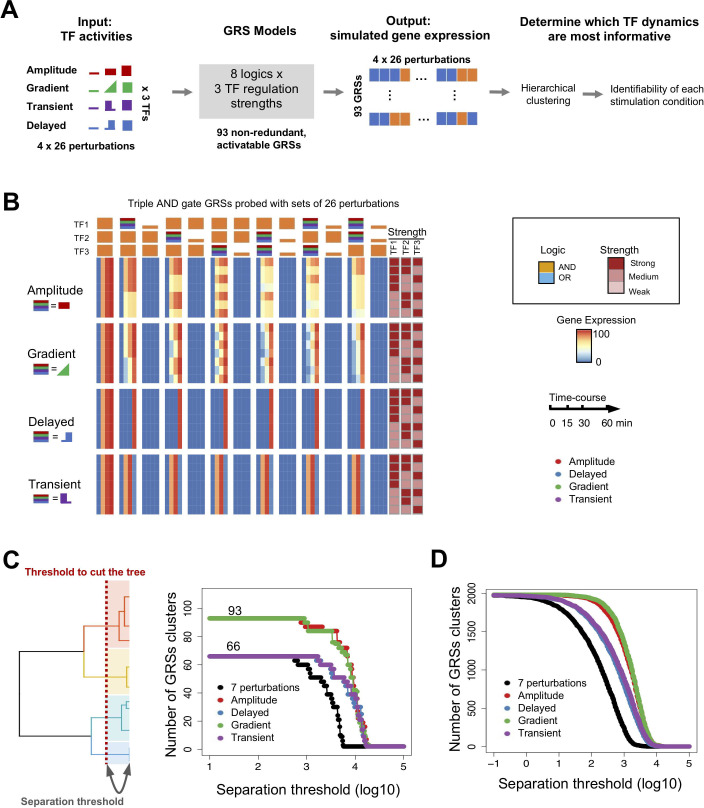
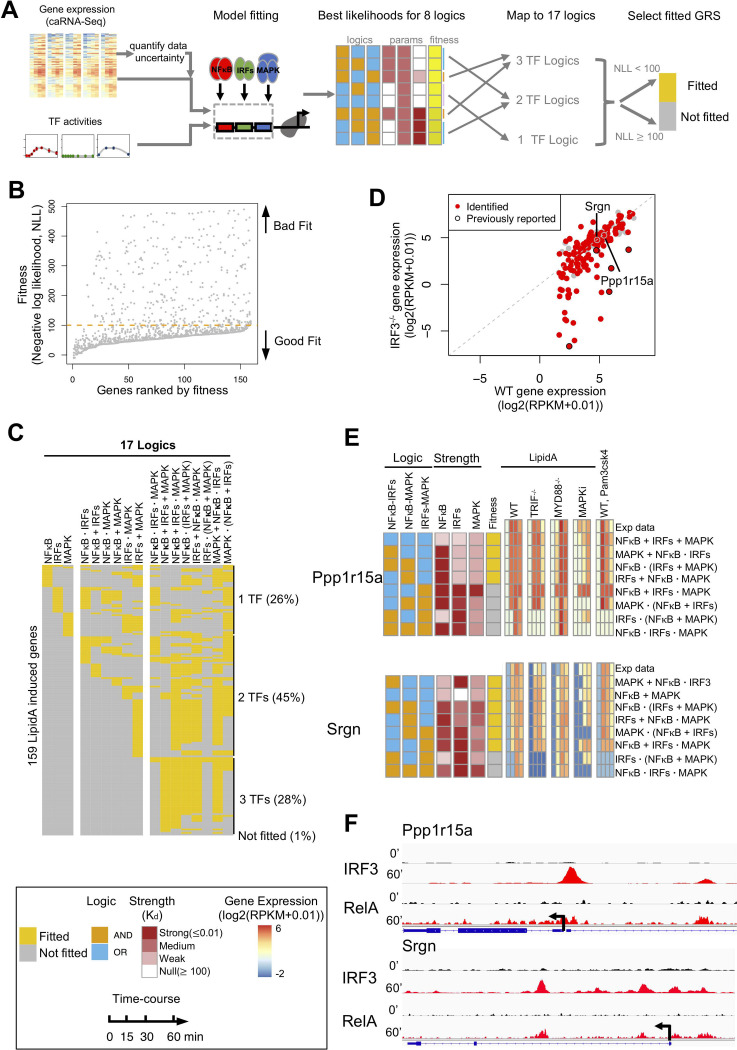
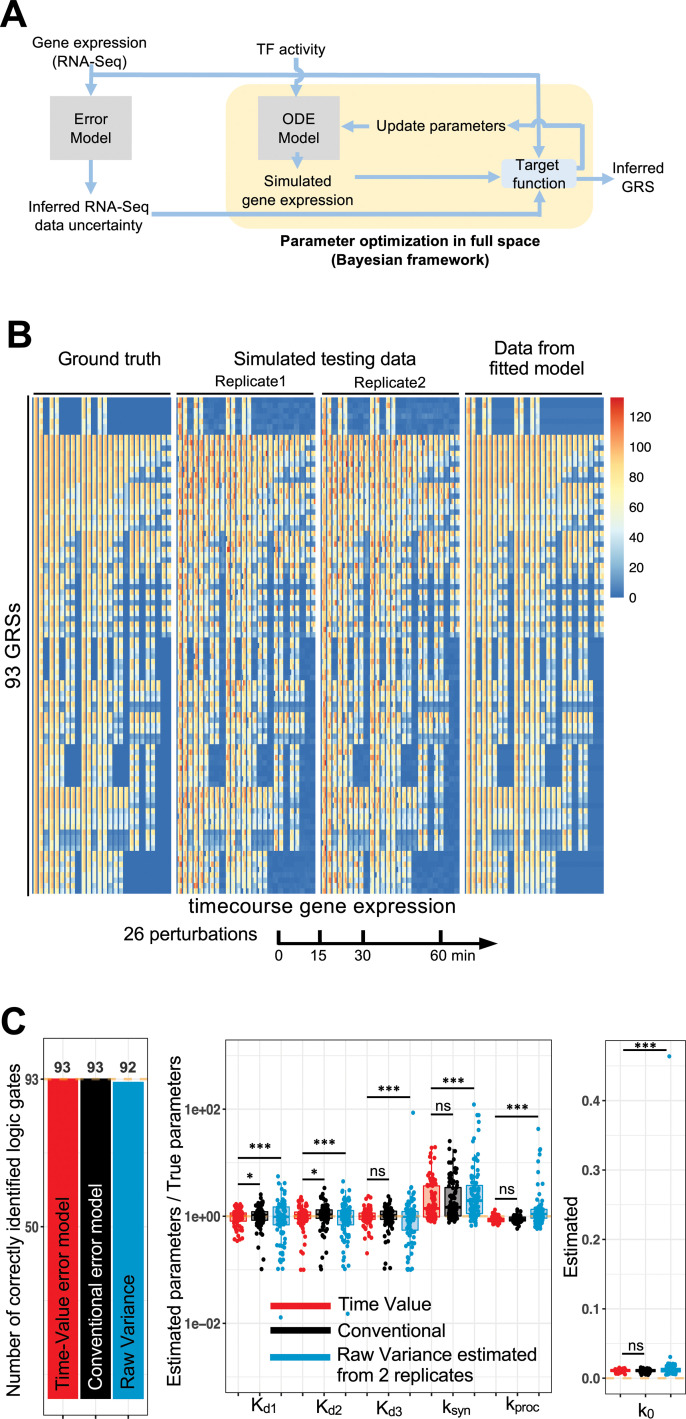
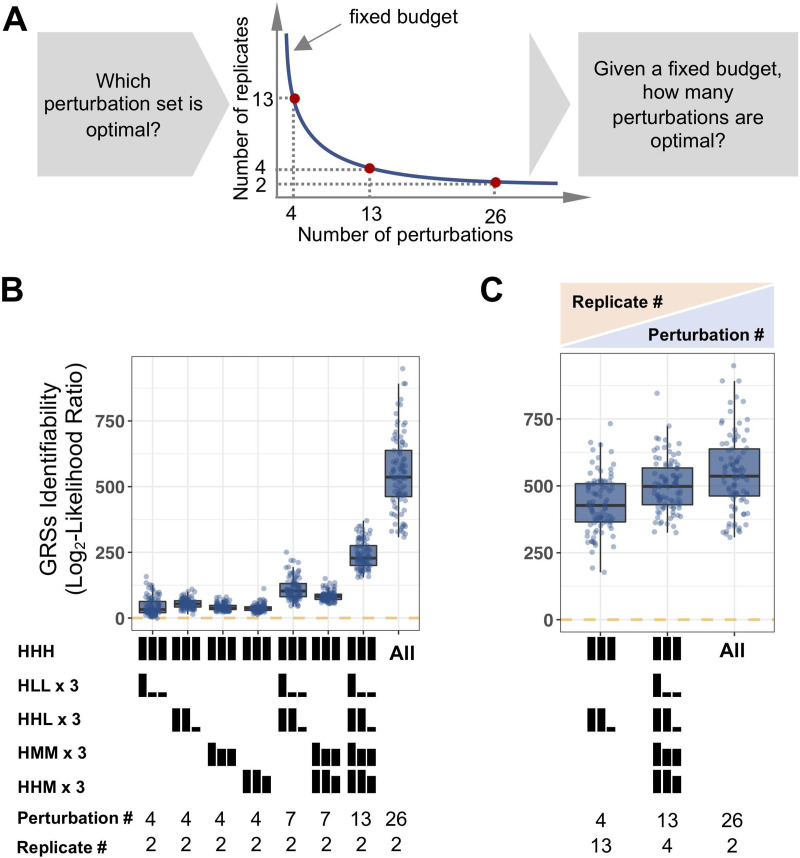
The effectiveness of immune responses depends on the precision of stimulus-responsive gene expression programs. Cells specify which genes to express by activating stimulus-specific combinations of stimulus-induced transcription factors (TFs). Their activities are decoded by a gene regulatory strategy (GRS) associated with each response gene. Here, we examined whether the GRSs of target genes may be inferred from stimulus-response (input-output) datasets, which remains an unresolved model-identifiability challenge. We developed a mechanistic modeling framework and computational workflow to determine the identifiability of all possible combinations of synergistic (AND) or non-synergistic (OR) GRSs involving three transcription factors. Considering different sets of perturbations for stimulus-response studies, we found that two thirds of GRSs are easily distinguishable but that substantially more quantitative data is required to distinguish the remaining third. To enhance the accuracy of the inference with timecourse experimental data, we developed an advanced error model that avoids error overestimates by distinguishing between value and temporal error. Incorporating this error model into a Bayesian framework, we show that GRS models can be identified for individual genes by considering multiple datasets. Our analysis rationalizes the allocation of experimental resources by identifying most informative TF stimulation conditions. Applying this computational workflow to experimental data of immune response genes in macrophages, we found that a much greater fraction of genes are combinatorially controlled than previously reported by considering compensation among transcription factors. Specifically, we revealed that a group of known NFκB target genes may also be regulated by IRF3, which is supported by chromatin immuno-precipitation analysis. Our study provides a computational workflow for designing and interpreting stimulus-response gene expression studies to identify underlying gene regulatory strategies and further a mechanistic understanding.
免疫反应的有效性取决于刺激反应性基因表达程序的精确性。细胞通过激活刺激特异性的刺激诱导转录因子(TF)组合来指定要表达的基因。它们的活性由与每个响应基因相关的基因调控策略(GRS)解码。在这里,我们研究了目标基因的 GRS 是否可以从刺激反应(输入-输出)数据集中推断出来,这仍然是一个未解决的模型可识别性挑战。我们开发了一种机械建模框架和计算工作流程,以确定涉及三个转录因子的协同(AND)或非协同(OR)GRS 的所有可能组合的可识别性。考虑到刺激反应研究的不同刺激数据集,我们发现三分之二的 GRS 很容易区分,但需要更多的定量数据来区分其余的三分之一。为了通过时间过程实验数据提高推断的准确性,我们开发了一种先进的误差模型,通过区分值和时间误差来避免误差高估。将此误差模型纳入贝叶斯框架中,我们表明通过考虑多个数据集,可以为单个基因识别 GRS 模型。我们的分析通过确定最具信息量的 TF 刺激条件来合理化实验资源的分配。将此计算工作流程应用于巨噬细胞中免疫反应基因的实验数据,我们发现,与以前的报告相比,通过考虑转录因子之间的补偿,组合控制的基因数量要多得多。具体来说,我们发现一组已知的 NFκB 靶基因也可能受到 IRF3 的调控,这得到了染色质免疫沉淀分析的支持。我们的研究提供了一种计算工作流程,用于设计和解释刺激反应基因表达研究,以识别潜在的基因调控策略,并进一步深入了解机制。