Saji Rinnu Sara, Prasana Johanan Christian, Muthu S, George Jacob
Department of Physics, Madras Christian College, East Tambaram 600059, Tamil Nadu, India.
University of Madras, Chennai, 600005, Tamil Nadu, India.
Heliyon. 2021 Jun 11;7(6):e07213. doi: 10.1016/j.heliyon.2021.e07213. eCollection 2021 Jun.
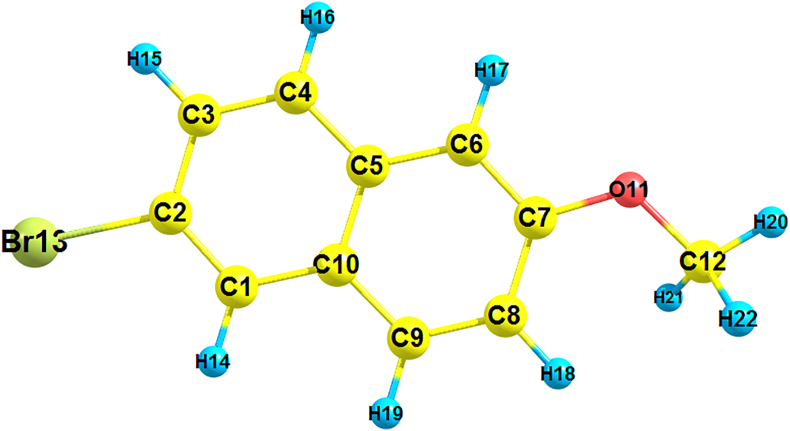
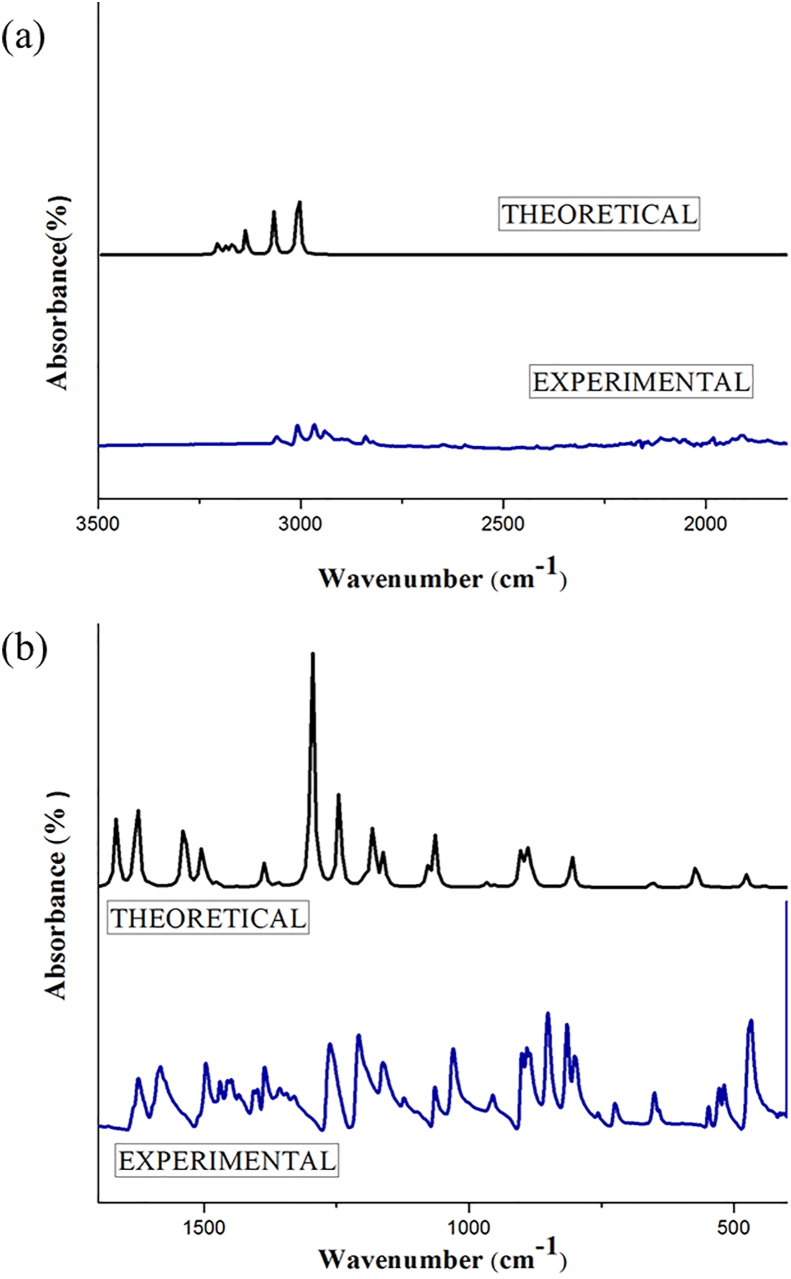
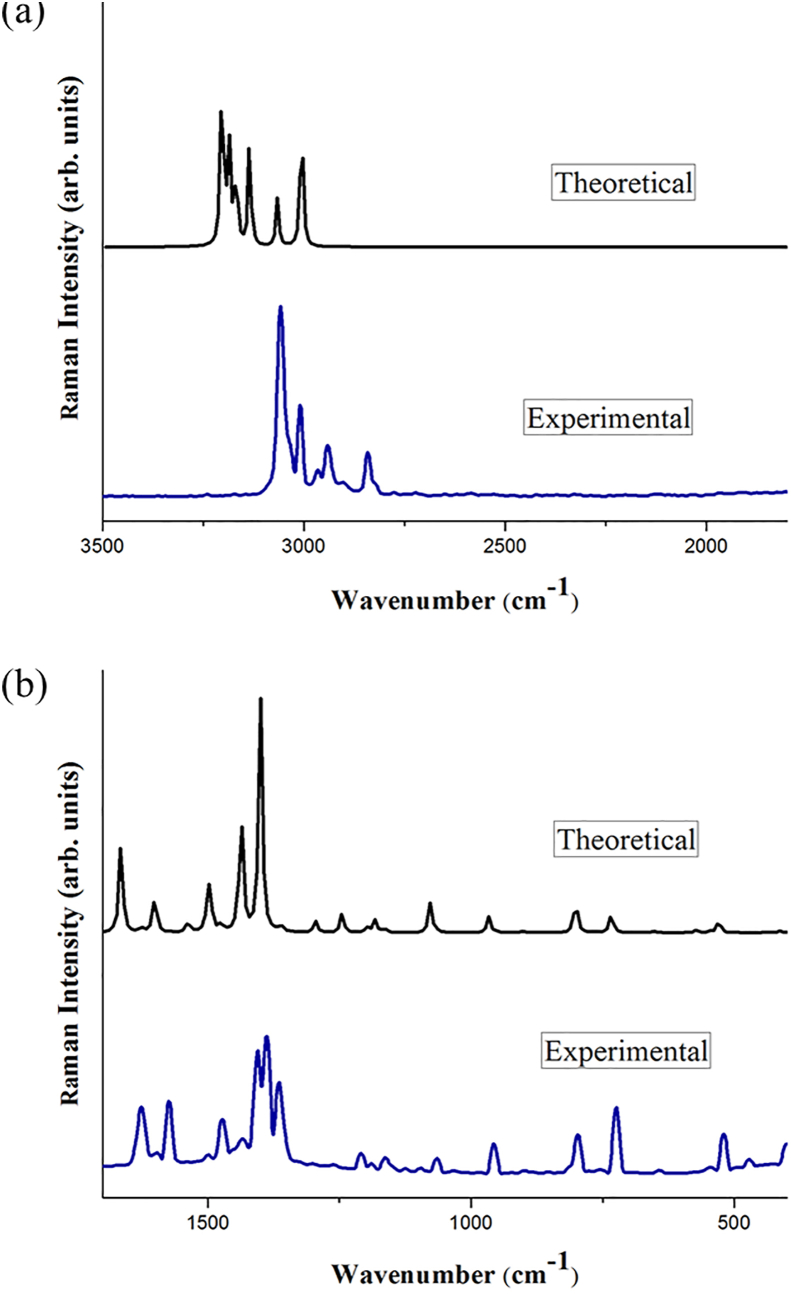
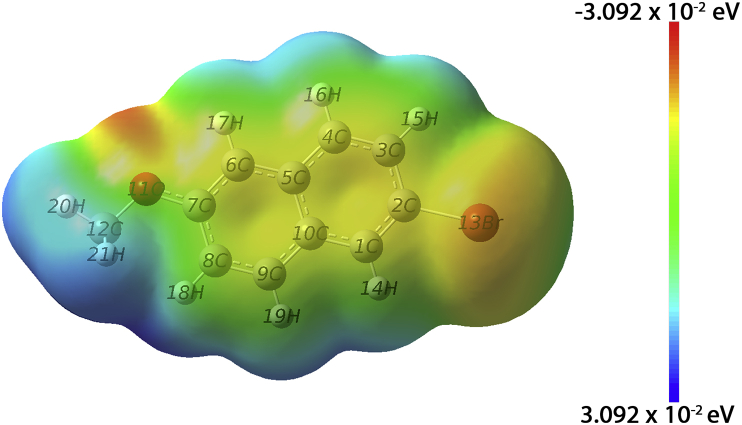
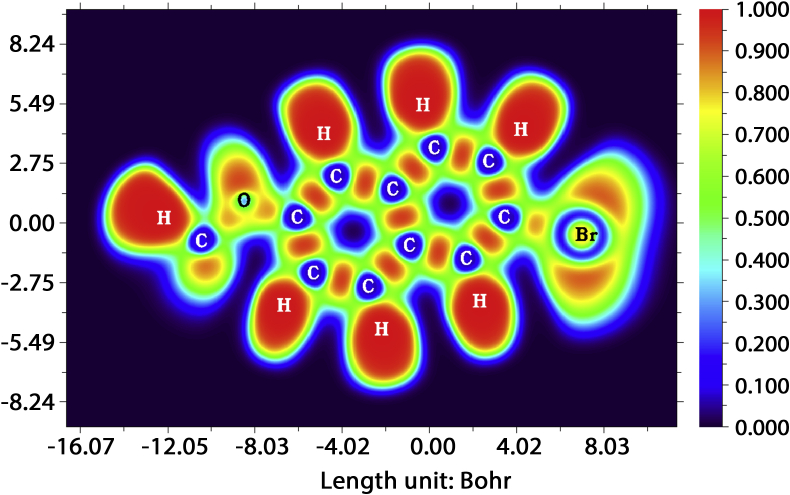
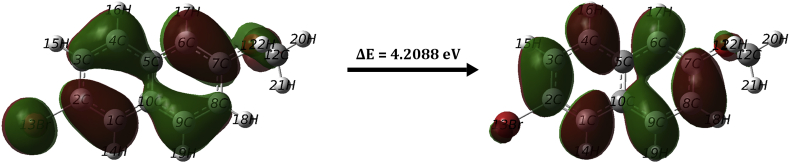
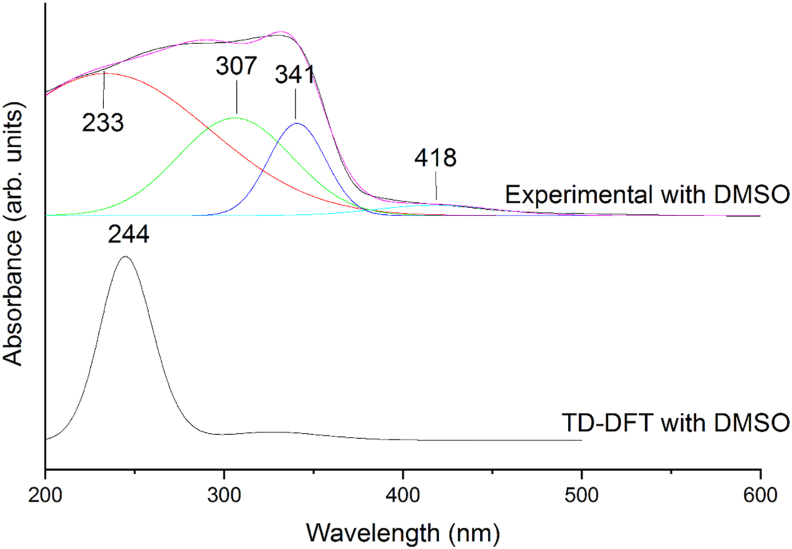
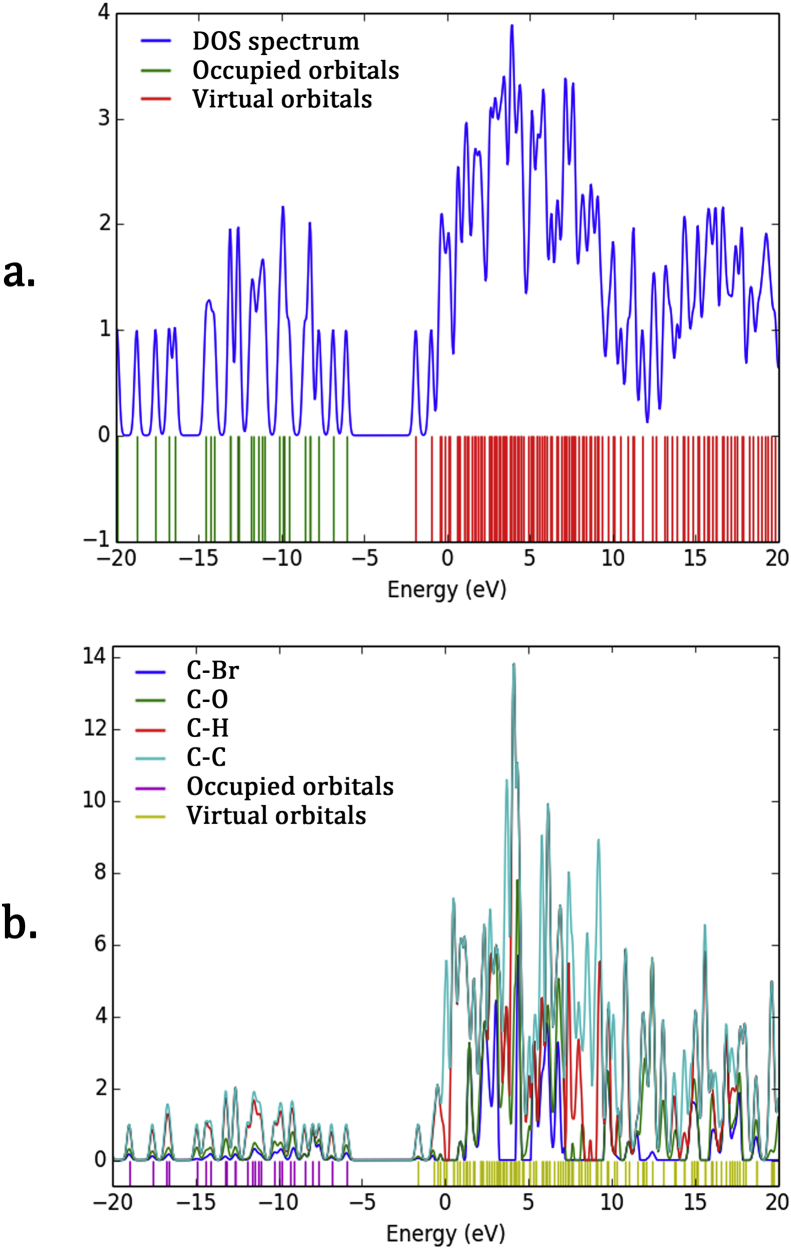
The vibrational, electronic and charge transfer studies on 2-bromo-6-methoxynaphthalene (2BMN) were done using DFT method with B3LYP/6-311++G(d,p) theory using GAUSSIAN 09W software. Theoretical and experimental investigations on FT-IR and FT Raman were executed on 2BMN. The calculated vibrational wavenumbers were scaled using suitable scaling factors and vibrational assignments were done to all modes of vibrations using Potential Energy Distribution (PED). Frontier Molecular Orbitals were calculated using TD-DFT method and the HOMO-LUMO energy gap was also obtained. Other electronic properties and global parameters for 2BMN were found using the HOMO-LUMO energy values. An energy gap of 4.208 eV shows the stability of the molecule. The reactive sites were predicted using Molecular Electrostatic Potential (MEP), Electron Localization Function (ELF) and Fukui calculations. Hence all electrophilic sites and nucleophilic areas of the molecule were determined. The delocalization of electron density was studied using NBO calculations. The intramolecular transitions and stability of structure were explained using in detail using the former. As the compound satisfies drug-like properties and has a softness value (indicating its less toxic nature), it may be used as a pharmaceutical product. Molecular docking studies were made and the protein-ligand binding properties were discussed. It was found out that title compound exhibits anti-cancer activities. The low binding energy predicts that the compound may be modified as a drug for treating Cancer.
使用GAUSSIAN 09W软件,采用B3LYP/6 - 311++G(d,p)理论的密度泛函理论(DFT)方法,对2 - 溴 - 6 - 甲氧基萘(2BMN)进行了振动、电子和电荷转移研究。对2BMN进行了傅里叶变换红外光谱(FT - IR)和傅里叶变换拉曼光谱的理论与实验研究。使用合适的标度因子对计算得到的振动波数进行标度,并利用势能分布(PED)对所有振动模式进行振动归属。采用含时密度泛函理论(TD - DFT)方法计算前线分子轨道,并得到最高占据分子轨道(HOMO) - 最低未占据分子轨道(LUMO)的能隙。利用HOMO - LUMO能量值发现了2BMN的其他电子性质和全局参数。4.208 eV的能隙表明该分子具有稳定性。使用分子静电势(MEP)、电子定域函数(ELF)和福井计算预测了反应位点。因此确定了分子的所有亲电位点和亲核区域。使用自然键轨道(NBO)计算研究了电子密度的离域。利用前者详细解释了分子内跃迁和结构稳定性。由于该化合物满足类药物性质且具有柔软度值(表明其低毒性),它可能用作药物产品。进行了分子对接研究并讨论了蛋白质 - 配体结合性质。发现标题化合物具有抗癌活性。低结合能预示该化合物可被修饰为治疗癌症的药物。