Hertzman Rebecca J, Deshpande Pooja, Leary Shay, Li Yueran, Ram Ramesh, Chopra Abha, Cooper Don, Watson Mark, Palubinsky Amy M, Mallal Simon, Gibson Andrew, Phillips Elizabeth J
Institute for Immunology and Infectious Diseases, Murdoch University, Murdoch, WA, Australia.
Department of Medicine, Vanderbilt University Medical Centre, Nashville, TN, United States.
Front Genet. 2021 Jun 17;12:642012. doi: 10.3389/fgene.2021.642012. eCollection 2021.
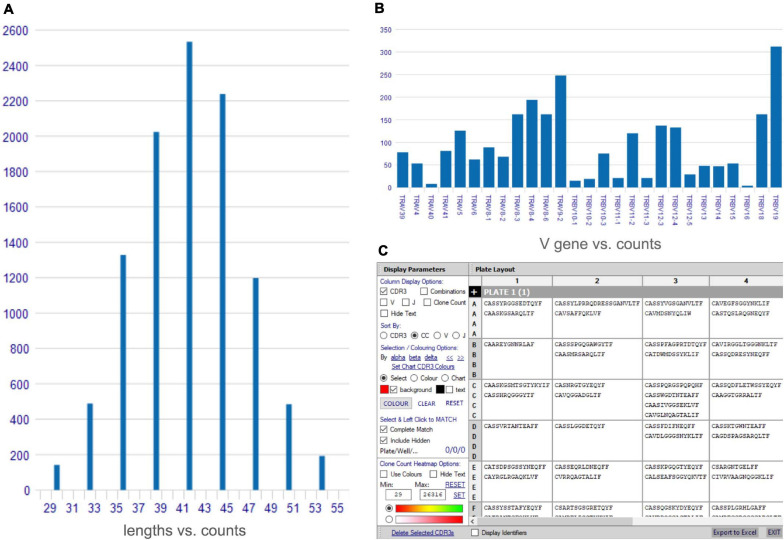
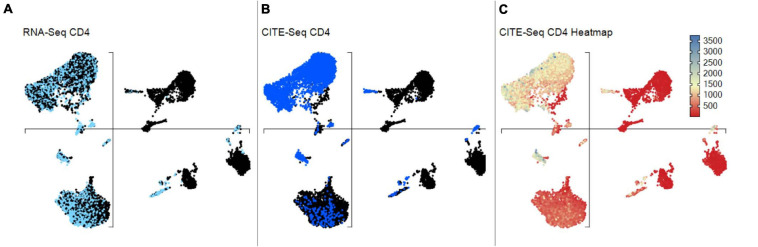
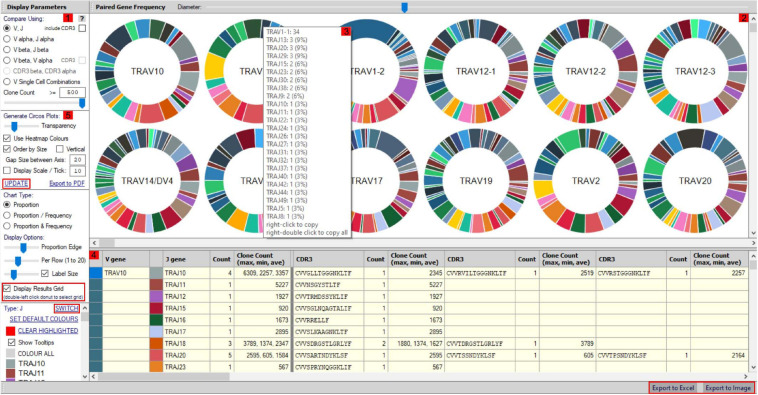
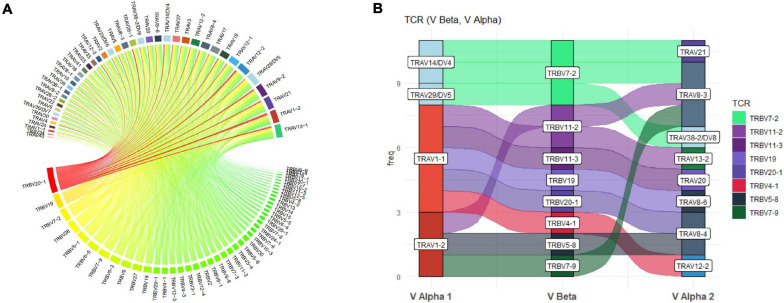
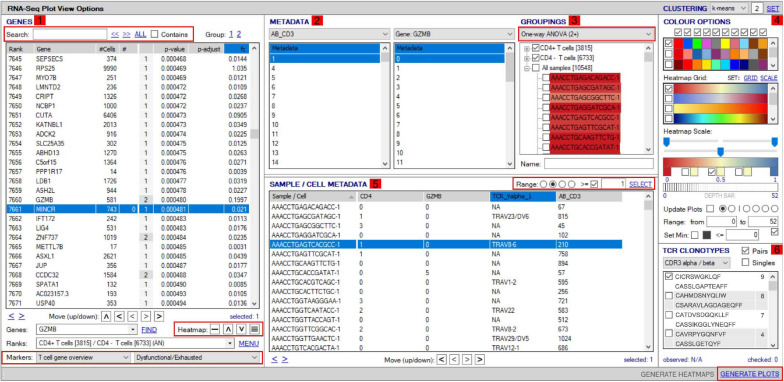
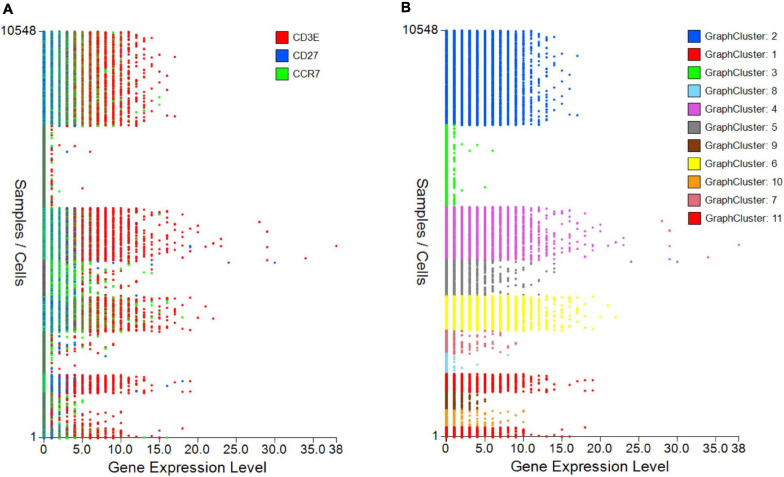
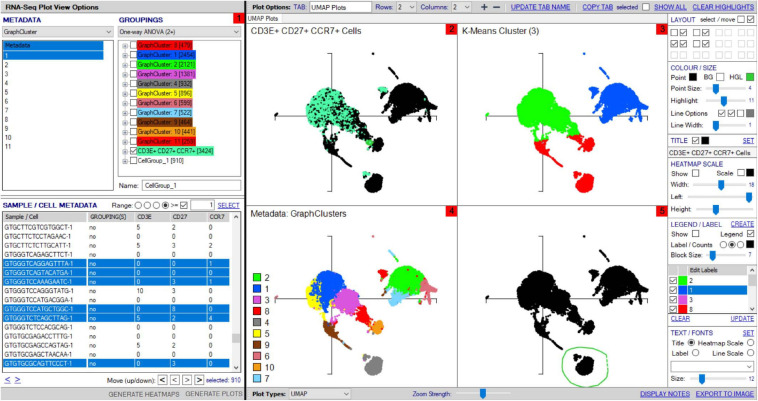
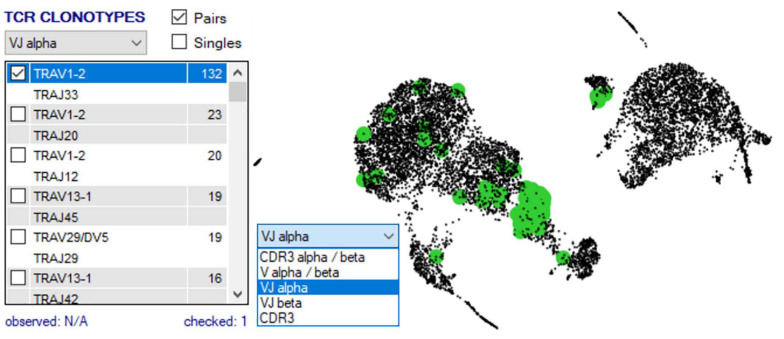
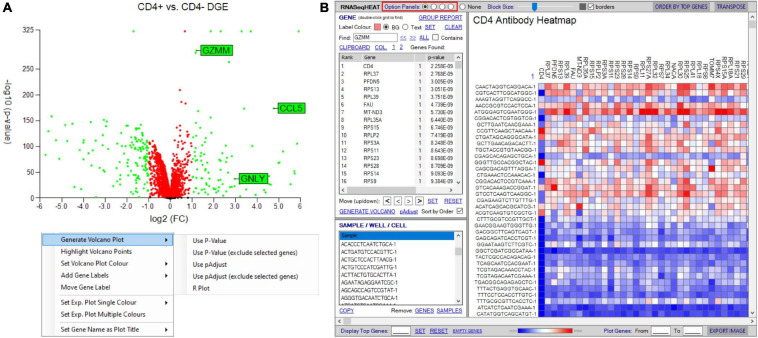
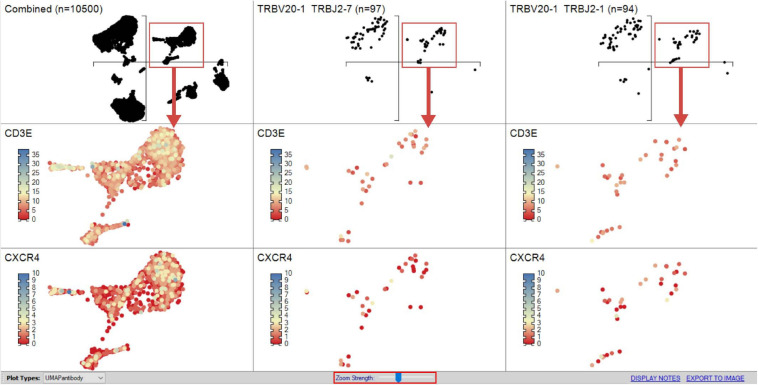
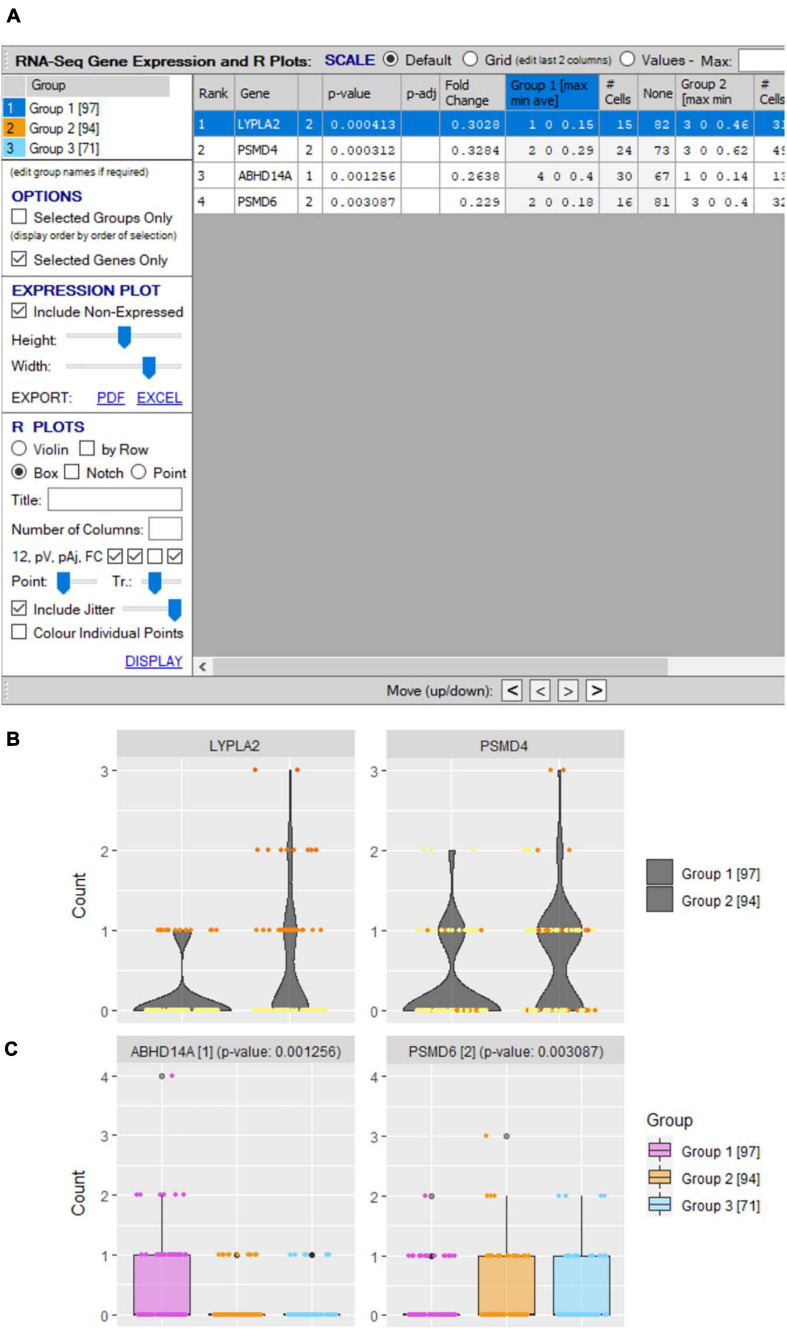
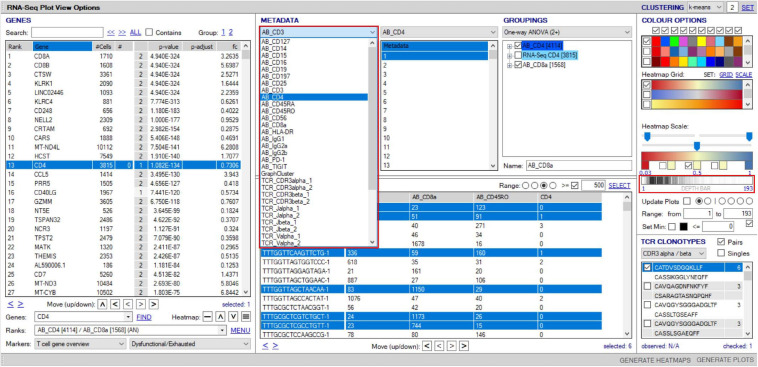
Type B adverse drug reactions (ADRs) are iatrogenic immune-mediated syndromes with mechanistic etiologies that remain incompletely understood. Some of the most severe ADRs, including delayed drug hypersensitivity reactions, are T-cell mediated, restricted by specific human leukocyte antigen risk alleles and sometimes by public or oligoclonal T-cell receptors (TCRs), central to the immunopathogenesis of tissue-damaging response. However, the specific cellular signatures of effector, regulatory, and accessory immune populations that mediate disease, define reaction phenotype, and determine severity have not been defined. Recent development of single-cell platforms bringing together advances in genomics and immunology provides the tools to simultaneously examine the full transcriptome, TCRs, and surface protein markers of highly heterogeneous immune cell populations at the site of the pathological response at a single-cell level. However, the requirement for advanced bioinformatics expertise and computational hardware and software has often limited the ability of investigators with the understanding of diseases and biological models to exploit these new approaches. Here we describe the features and use of a state-of-the-art, fully integrated application for analysis and visualization of multiomic single-cell data called Visual Genomics Analysis Studio (VGAS). This unique user-friendly, Windows-based graphical user interface is specifically designed to enable investigators to interrogate their own data. While VGAS also includes tools for sequence alignment and identification of associations with host or organism genetic polymorphisms, in this review we focus on its application for analysis of single-cell TCR-RNA-Cellular Indexing of Transcriptomes and Epitopes by Sequencing (CITE)-seq, enabling holistic cellular characterization by unbiased transcriptome and select surface proteome. Critically, VGAS does not require user-directed coding or access to high-performance computers, instead incorporating performance-optimized hidden code to provide application-based fast and intuitive tools for data analyses and production of high-resolution publication-ready graphics on standard specification laptops. Specifically, it allows analyses of comprehensive single-cell TCR sequencing (scTCR-seq) data, detailing (i) functional pairings of α-β heterodimer TCRs, (ii) one-click histograms to display entropy and gene rearrangements, and (iii) Circos and Sankey plots to visualize clonality and dominance. For unbiased single-cell RNA sequencing (scRNA-seq) analyses, users extract cell transcriptome signatures according to global structure via principal component analysis, t-distributed stochastic neighborhood embedding, or uniform manifold approximation and projection plots, with overlay of scTCR-seq enabling identification and selection of the immunodominant TCR-expressing populations. Further integration with similar sequence-based detection of surface protein markers using oligo-labeled antibodies (CITE-seq) provides comparative understanding of surface protein expression, with differential gene or protein analyses visualized using volcano plot or heatmap functions. These data can be compared to reference cell atlases or suitable controls to reveal discrete disease-specific subsets, from epithelial to tissue-resident memory T-cells, and activation status, from senescence through exhaustion, with more finite transcript expression displayed as violin and box plots. Importantly, guided tutorial videos are available, as are regular application updates based on the latest advances in bioinformatics and user feedback.
B型药物不良反应(ADR)是医源性免疫介导综合征,其发病机制尚未完全明确。一些最严重的ADR,包括迟发性药物超敏反应,是由T细胞介导的,受特定人类白细胞抗原风险等位基因限制,有时也受公共或寡克隆T细胞受体(TCR)限制,这是组织损伤反应免疫发病机制的核心。然而,介导疾病、定义反应表型并决定严重程度的效应、调节和辅助免疫群体的特定细胞特征尚未明确。单细胞平台的最新发展结合了基因组学和免疫学的进展,提供了在单细胞水平上同时检查病理反应部位高度异质免疫细胞群体的完整转录组、TCR和表面蛋白标志物的工具。然而,对先进生物信息学专业知识以及计算硬件和软件的需求常常限制了对疾病和生物学模型有理解的研究人员利用这些新方法的能力。在此,我们描述了一种最先进的、用于多组学单细胞数据的分析和可视化的完全集成应用程序——视觉基因组分析工作室(VGAS)的特点和用途。这个独特的、用户友好的、基于Windows的图形用户界面专门设计用于使研究人员能够查询自己的数据。虽然VGAS还包括序列比对以及与宿主或生物体基因多态性关联识别的工具,但在本综述中,我们重点关注其在分析单细胞TCR - RNA - 转录组和表位测序细胞索引(CITE)- seq方面的应用,通过无偏转录组和选定的表面蛋白质组实现整体细胞表征。至关重要的是,VGAS不需要用户直接编码或访问高性能计算机,而是纳入了性能优化的隐藏代码,以在标准规格笔记本电脑上提供基于应用程序的快速且直观的数据分析工具以及用于生成高分辨率可用于发表的图形。具体而言,它允许对全面的单细胞TCR测序(scTCR - seq)数据进行分析,详细说明(i)α - β异二聚体TCR的功能配对,(ii)一键式直方图以显示熵和基因重排,以及(iii)Circos图和桑基图以可视化克隆性和优势度。对于无偏单细胞RNA测序(scRNA - seq)分析,用户通过主成分分析、t分布随机邻域嵌入或均匀流形近似与投影图根据全局结构提取细胞转录组特征,并叠加scTCR - seq能够识别和选择表达免疫显性TCR的群体。使用寡核苷酸标记抗体对表面蛋白标志物进行类似基于序列的检测(CITE - seq)的进一步整合,提供了对表面蛋白表达的比较理解,使用火山图或热图功能可视化差异基因或蛋白分析。这些数据可以与参考细胞图谱或合适的对照进行比较,以揭示从上皮细胞到组织驻留记忆T细胞的离散疾病特异性亚群,以及从衰老到耗竭的激活状态,以小提琴图和箱线图显示更有限的转录表达。重要的是,有指导教程视频可用,并且会根据生物信息学的最新进展和用户反馈定期进行应用程序更新。