Murithi James M, Deni Ioanna, Pasaje Charisse Flerida A, Okombo John, Bridgford Jessica L, Gnädig Nina F, Edwards Rachel L, Yeo Tomas, Mok Sachel, Burkhard Anna Y, Coburn-Flynn Olivia, Istvan Eva S, Sakata-Kato Tomoyo, Gomez-Lorenzo Maria G, Cowell Annie N, Wicht Kathryn J, Le Manach Claire, Kalantarov Gavreel F, Dey Sumanta, Duffey Maëlle, Laleu Benoît, Lukens Amanda K, Ottilie Sabine, Vanaerschot Manu, Trakht Ilya N, Gamo Francisco-Javier, Wirth Dyann F, Goldberg Daniel E, Odom John Audrey R, Chibale Kelly, Winzeler Elizabeth A, Niles Jacquin C, Fidock David A
Department of Microbiology and Immunology, Columbia University Irving Medical Center, New York, NY 10032, USA.
Department of Biological Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA.
Cell Chem Biol. 2022 May 19;29(5):824-839.e6. doi: 10.1016/j.chembiol.2021.06.006. Epub 2021 Jul 6.
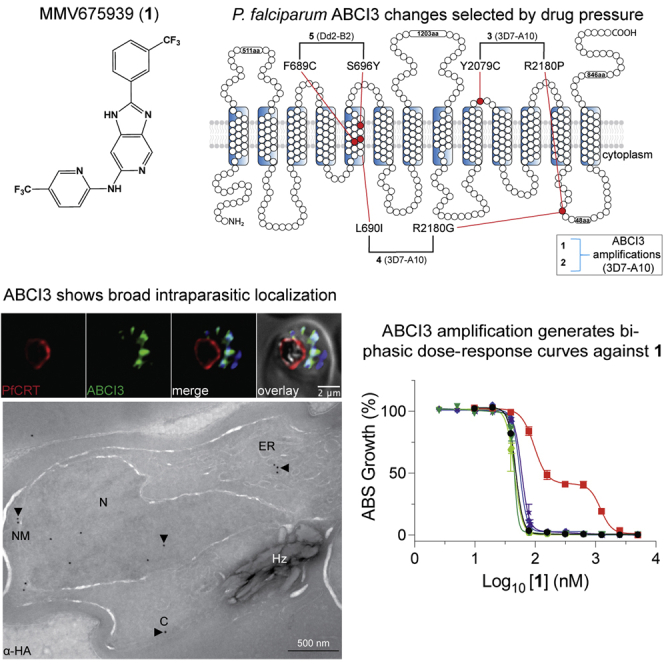
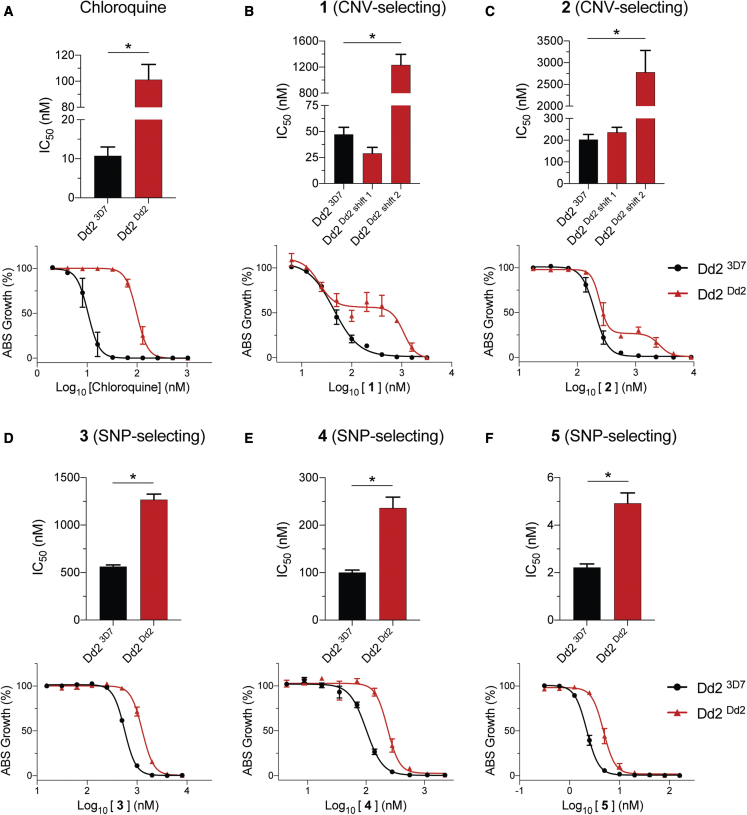
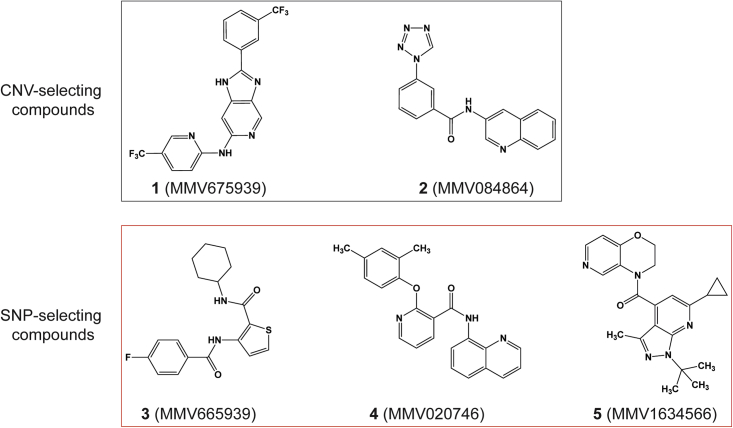
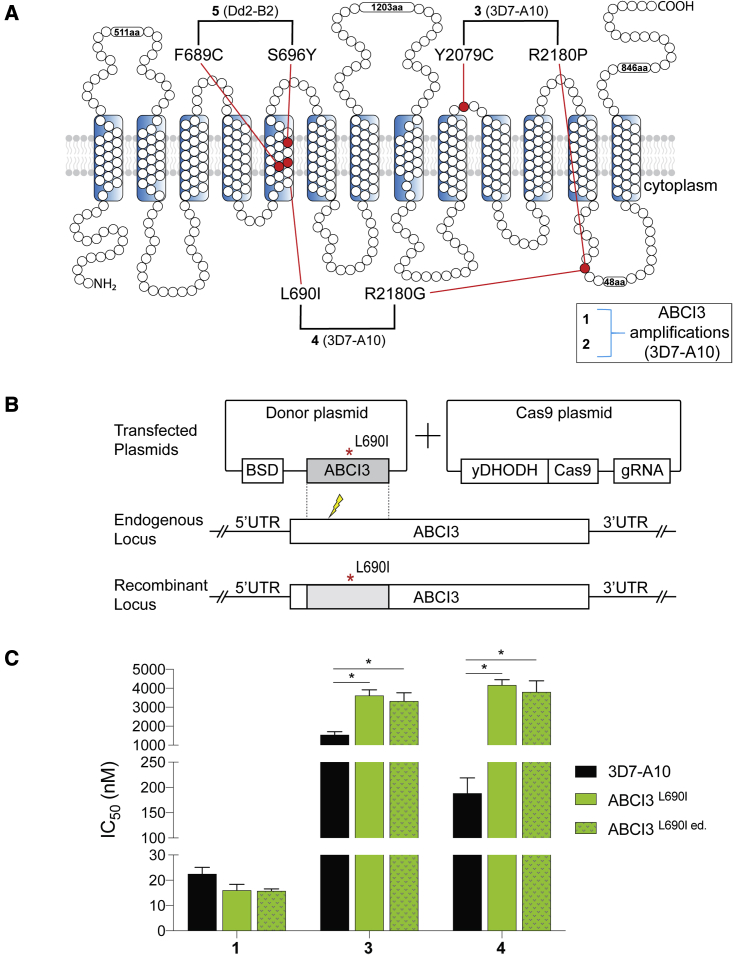
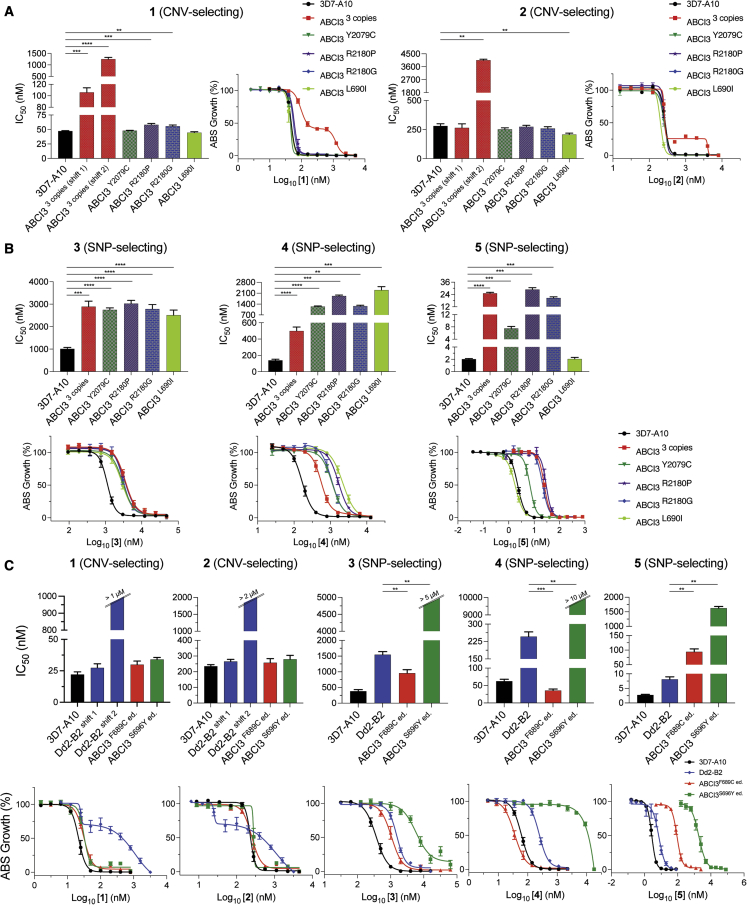
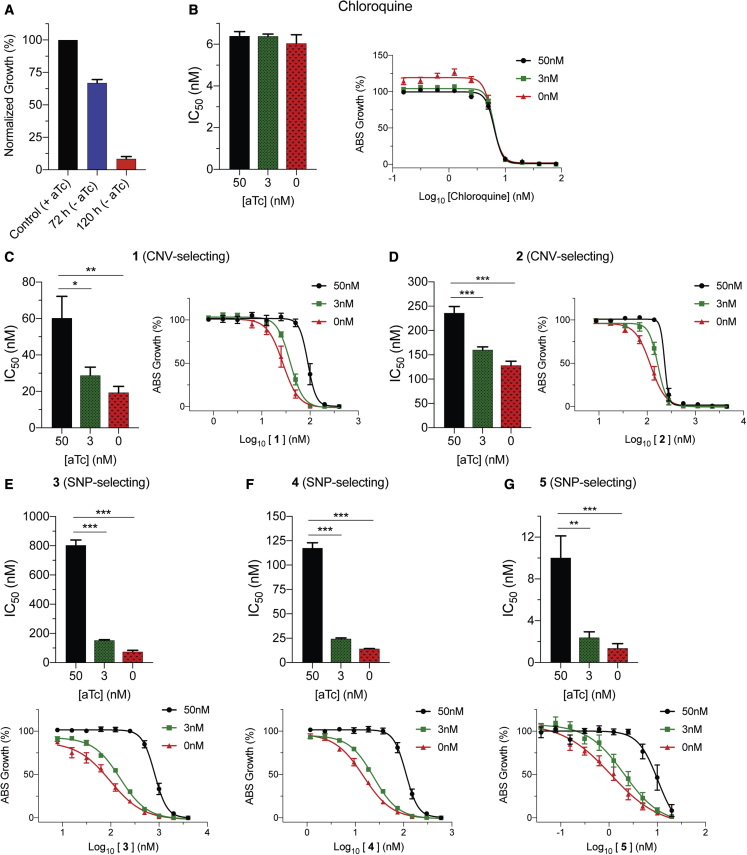
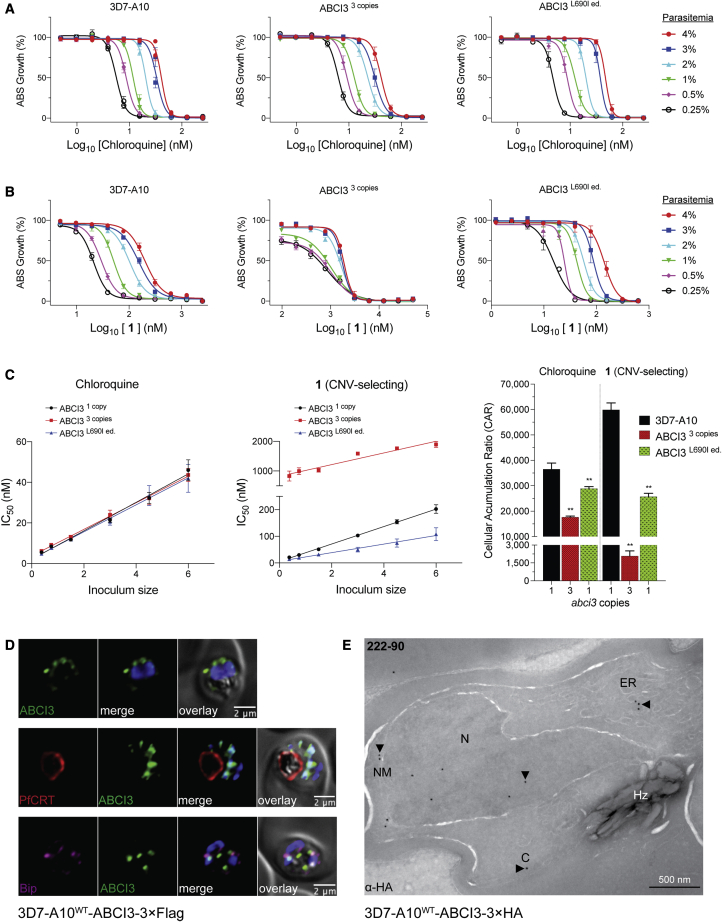
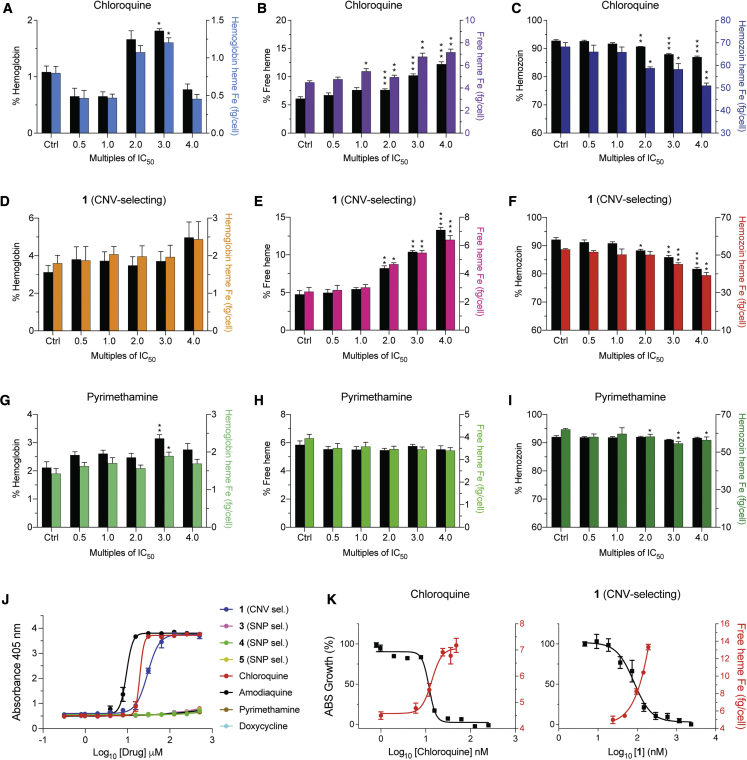
Widespread Plasmodium falciparum resistance to first-line antimalarials underscores the vital need to develop compounds with novel modes of action and identify new druggable targets. Here, we profile five compounds that potently inhibit P. falciparum asexual blood stages. Resistance selection studies with three carboxamide-containing compounds, confirmed by gene editing and conditional knockdowns, identify point mutations in the parasite transporter ABCI3 as the primary mediator of resistance. Selection studies with imidazopyridine or quinoline-carboxamide compounds also yield changes in ABCI3, this time through gene amplification. Imidazopyridine mode of action is attributed to inhibition of heme detoxification, as evidenced by cellular accumulation and heme fractionation assays. For the copy-number variation-selecting imidazopyridine and quinoline-carboxamide compounds, we find that resistance, manifesting as a biphasic concentration-response curve, can independently be mediated by mutations in the chloroquine resistance transporter PfCRT. These studies reveal the interconnectedness of P. falciparum transporters in overcoming drug pressure in different parasite strains.
恶性疟原虫对一线抗疟药物的广泛耐药性凸显了开发具有新作用模式的化合物并确定新的可成药靶点的迫切需求。在此,我们对五种能有效抑制恶性疟原虫无性血液阶段的化合物进行了分析。对三种含羧酰胺化合物的耐药性选择研究,经基因编辑和条件性敲低证实,确定寄生虫转运蛋白ABCI3中的点突变是耐药性的主要介导因素。对咪唑并吡啶或喹啉 - 羧酰胺化合物的选择研究也导致ABCI3发生变化,这次是通过基因扩增。咪唑并吡啶的作用模式归因于对血红素解毒的抑制,细胞积累和血红素分级分析证明了这一点。对于拷贝数变异选择的咪唑并吡啶和喹啉 - 羧酰胺化合物,我们发现耐药性表现为双相浓度 - 反应曲线,可独立地由氯喹耐药转运蛋白PfCRT中的突变介导。这些研究揭示了恶性疟原虫转运蛋白在克服不同寄生虫菌株的药物压力方面的相互联系。