Gopi Soundhararajan, Aranganathan Akashnathan, Naganathan Athi N
Department of Biotechnology, Bhupat & Jyoti Mehta School of Biosciences, Indian Institute of Technology Madras, Chennai 600036, India.
Curr Res Struct Biol. 2019 Oct 23;1:6-12. doi: 10.1016/j.crstbi.2019.10.002. eCollection 2019 Nov.
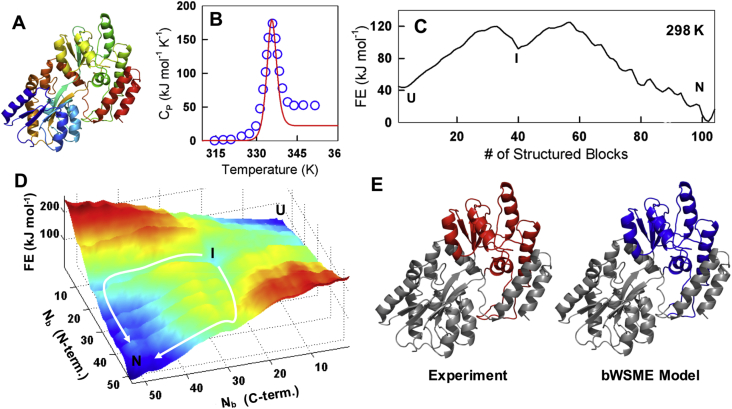
Statistical mechanical models that afford an intermediate resolution between macroscopic chemical models and all-atom simulations have been successful in capturing folding behaviors of many small single-domain proteins. However, the applicability of one such successful approach, the Wako-Saitô-Muñoz-Eaton (WSME) model, is limited by the size of the protein as the number of conformations grows exponentially with protein length. In this work, we surmount this size limitation by introducing a novel approximation that treats stretches of 3 or 4 residues as blocks, thus reducing the phase space by nearly three orders of magnitude. The performance of the 'bWSME' model is validated by comparing the predictions for a globular enzyme (RNase H) and a repeat protein (IκBα), against experimental observables and the model without block approximation. Finally, as a proof of concept, we predict the free-energy surface of the 370-residue, multi-domain maltose binding protein and identify an intermediate in good agreement with single-molecule force-spectroscopy measurements. The bWSME model can thus be employed as a quantitative predictive tool to explore the conformational landscapes of large proteins, extract the structural features of putative intermediates, identify parallel folding paths, and thus aid in the interpretation of both ensemble and single-molecule experiments.
统计力学模型能够在宏观化学模型和全原子模拟之间提供中等分辨率,已成功捕捉到许多小单域蛋白质的折叠行为。然而,一种成功的方法——和光-斋藤-穆尼奥斯-伊顿(WSME)模型,其适用性受到蛋白质大小的限制,因为构象数量会随着蛋白质长度呈指数增长。在这项工作中,我们通过引入一种新的近似方法克服了这一大小限制,该方法将3个或4个残基的片段视为块,从而将相空间减少了近三个数量级。通过将球状酶(核糖核酸酶H)和重复蛋白(IκBα)的预测结果与实验观测值以及无块近似模型进行比较,验证了“bWSME”模型的性能。最后,作为概念验证,我们预测了370个残基的多域麦芽糖结合蛋白的自由能表面,并确定了一个与单分子力谱测量结果高度一致的中间体。因此,bWSME模型可作为一种定量预测工具,用于探索大蛋白质的构象景观,提取假定中间体的结构特征,识别平行折叠路径,从而有助于解释整体实验和单分子实验。