Department of Molecular and Cell Biology, University of Connecticut, Storrs, CT 06268-3125, USA.
Bioinformatics Institute, School of Biological Sciences, The University of Auckland, Auckland 1010, New Zealand.
Syst Biol. 2022 Feb 10;71(2):396-409. doi: 10.1093/sysbio/syab060.
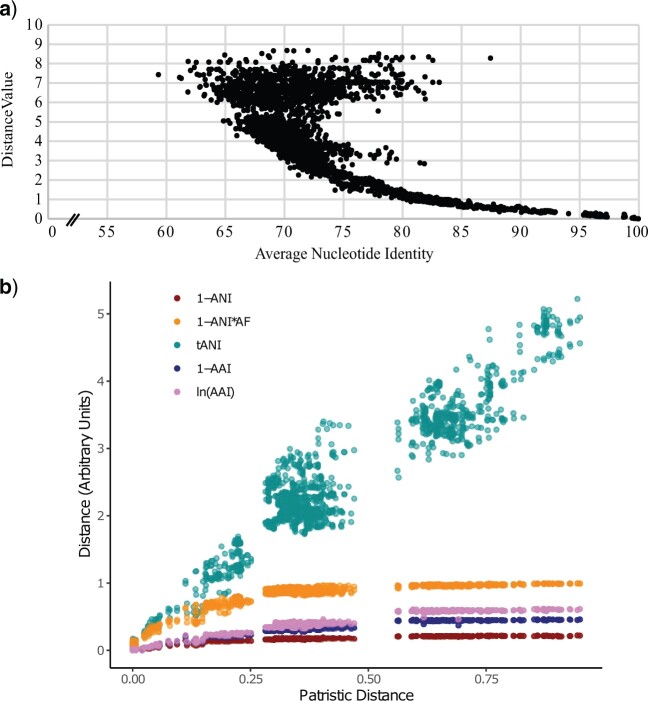
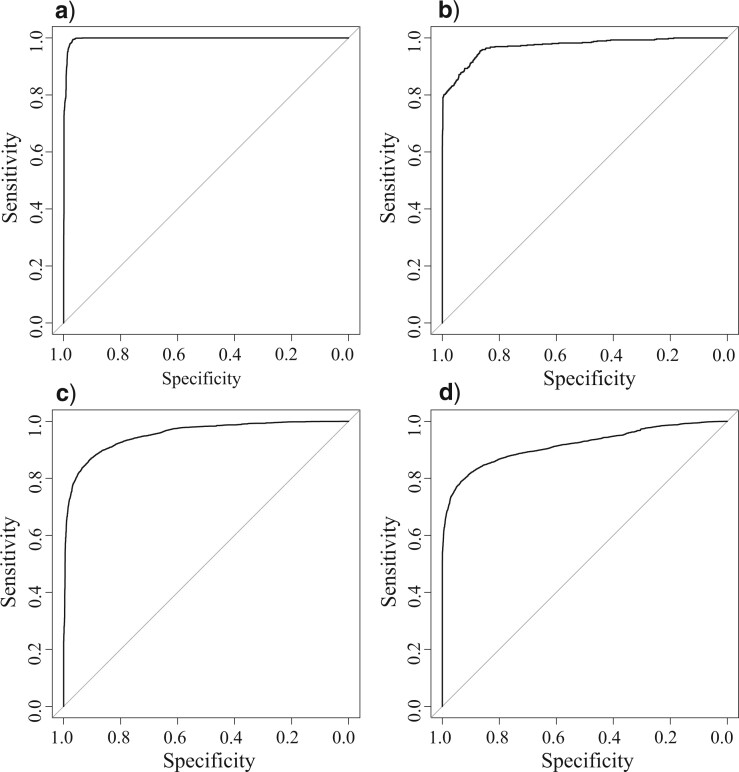
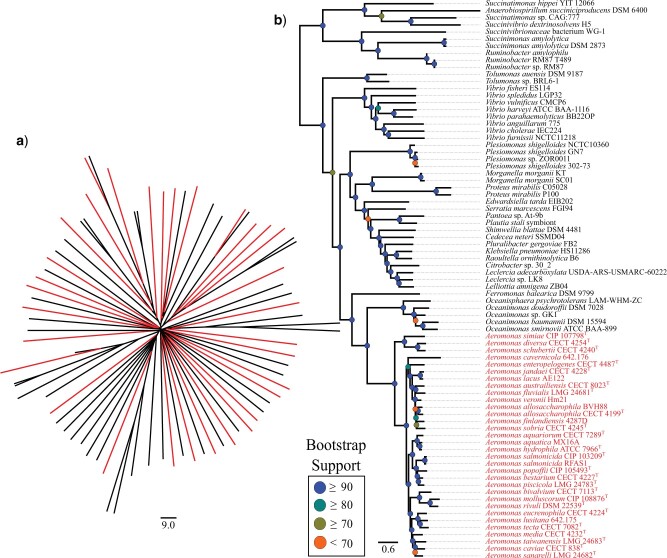
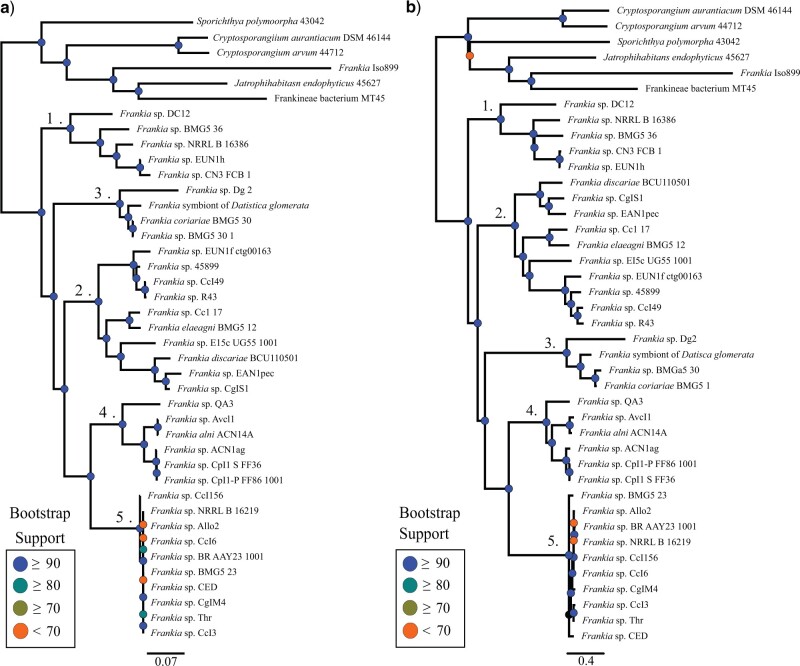
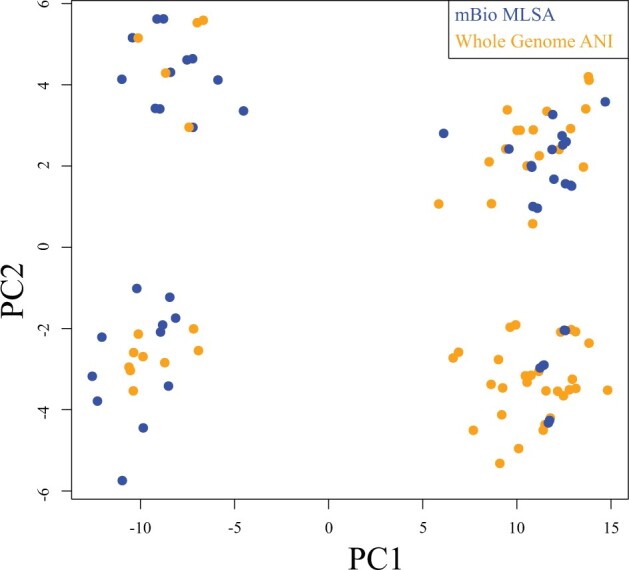
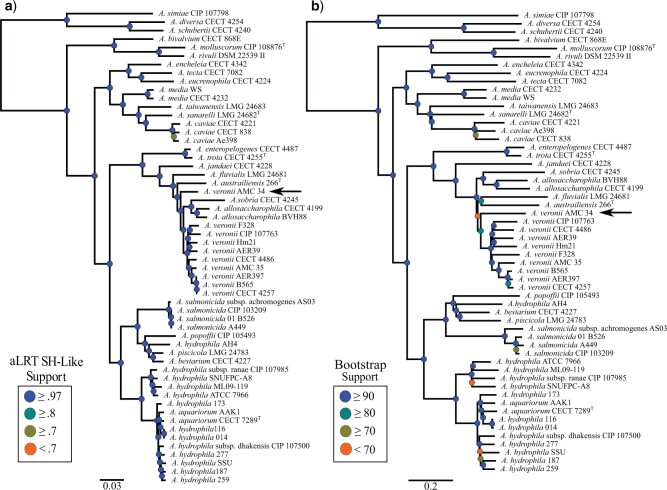
Whole-genome comparisons based on average nucleotide identities (ANI) and the genome-to-genome distance calculator have risen to prominence in rapidly classifying prokaryotic taxa using whole-genome sequences. Some implementations have even been proposed as a new standard in species classification and have become a common technique for papers describing newly sequenced genomes. However, attempts to apply whole-genome divergence data to the delineation of higher taxonomic units and to phylogenetic inference have had difficulty matching those produced by more complex phylogenetic methods. We present a novel method for generating statistically supported phylogenies of archaeal and bacterial groups using a combined ANI and alignment fraction-based metric. For the test cases to which we applied the developed approach, we obtained results comparable with other methodologies up to at least the family level. The developed method uses nonparametric bootstrapping to gauge support for inferred groups. This method offers the opportunity to make use of whole-genome comparison data, that is already being generated, to quickly produce phylogenies including support for inferred groups. Additionally, the developed ANI methodology can assist the classification of higher taxonomic groups.[Average nucleotide identity (ANI); genome evolution; prokaryotic species delineation; taxonomy.].
基于平均核苷酸同一性 (ANI) 和基因组到基因组距离计算器的全基因组比较在使用全基因组序列快速分类原核分类群方面引起了关注。一些实现甚至被提议作为物种分类的新标准,并成为描述新测序基因组的论文的常用技术。然而,将全基因组分歧数据应用于更高分类单元的划分和系统发育推断的尝试,很难与更复杂的系统发育方法产生的结果相匹配。我们提出了一种使用组合 ANI 和基于比对片段的度量来生成基于全基因组比较数据的古菌和细菌类群的统计支持系统发育的新方法。对于我们应用所开发方法的测试案例,我们获得的结果至少与家族级别一样可与其他方法学相媲美。所开发的方法使用非参数自举来衡量推断组的支持。该方法提供了利用已经生成的全基因组比较数据快速生成包括推断组支持的系统发育的机会。此外,所开发的 ANI 方法学可有助于对更高分类群的分类。[平均核苷酸同一性 (ANI); 基因组进化; 原核物种划分; 分类学。]