Vale Filipa F, Vítor Jorge M B, Marques Andreia T, Azevedo-Pereira José Miguel, Anes Elsa, Goncalves Joao
Pathogen Genome Bioinformatics and Computational Biology, Research Institute for Medicines (iMed-ULisboa), Faculty of Pharmacy, Universidade de Lisboa, Lisboa 1649-003, Portugal.
Pathogen Genome Bioinformatics and Computational Biology, Research Institute for Medicines (iMed-ULisboa), Faculty of Pharmacy, Universidade de Lisboa, Lisboa 1649-003, Portugal; Pharmacy, Pharmacology and Health Technologies Department, Faculty of Pharmacy, Universidade de Lisboa, Lisbon 1649-003, Portugal.
Virus Res. 2021 Oct 15;304:198526. doi: 10.1016/j.virusres.2021.198526. Epub 2021 Jul 30.
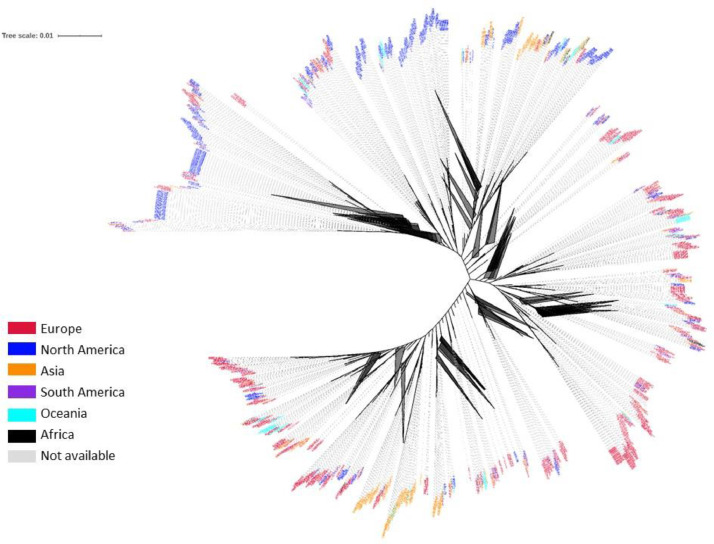
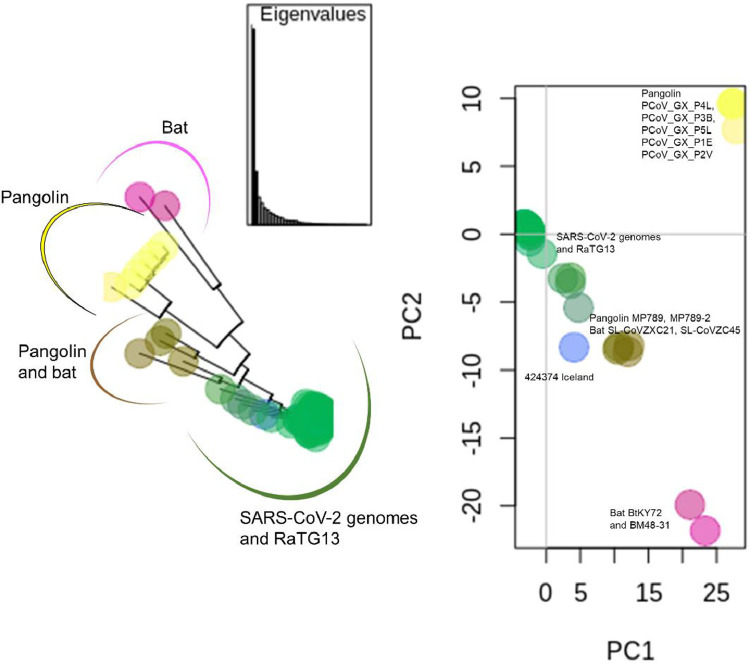
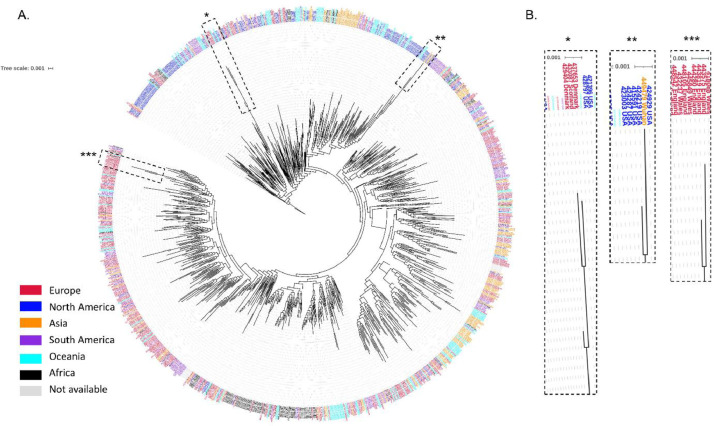
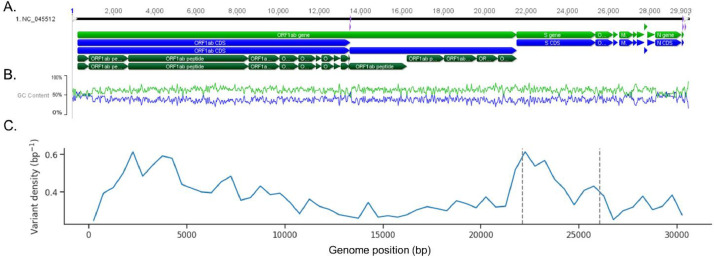
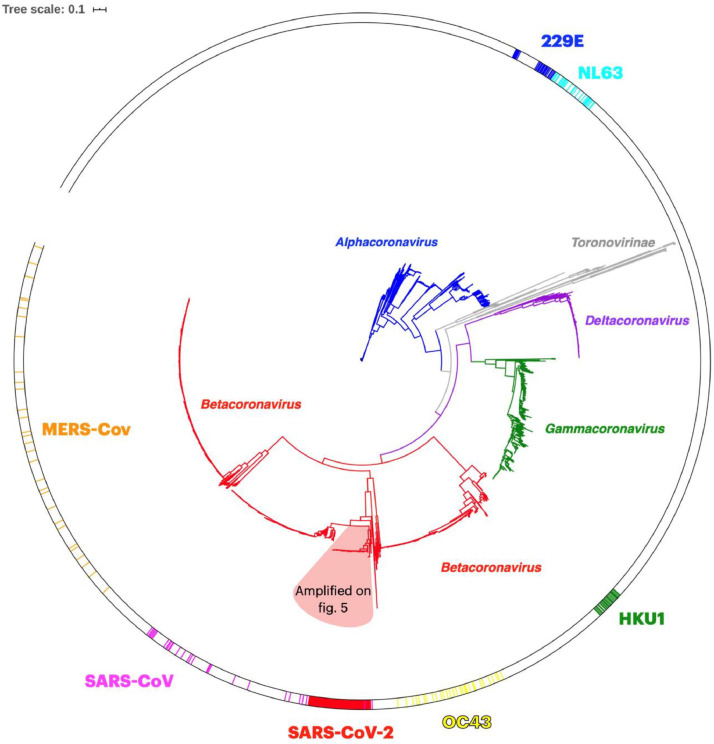
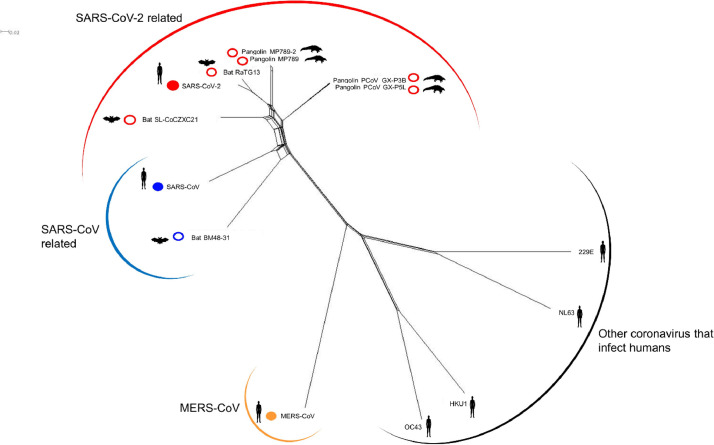
The coronavirus disease 2019 (COVID-19) pandemic caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) poses innumerous challenges, like understanding what triggered the emergence of this new human virus, how this RNA virus is evolving or how the variability of viral genome may impact the primary structure of proteins that are targets for vaccine. We analyzed 19471 SARS-CoV-2 genomes available at the GISAID database from all over the world and 3335 genomes of other Coronoviridae family members available at GenBank, collecting SARS-CoV-2 high-quality genomes and distinct Coronoviridae family genomes. Additionally, we analyzed 199,984 spike glycoprotein sequences. Here, we identify a SARS-CoV-2 emerging cluster containing 13 closely related genomes isolated from bat and pangolin that showed evidence of recombination, which may have contributed to the emergence of SARS-CoV-2. The analyzed SARS-CoV-2 genomes presented 9632 single nucleotide variants (SNVs) corresponding to a variant density of 0.3 over the genome, and a clear geographic distribution. SNVs are unevenly distributed throughout the genome and hotspots for mutations were found for the spike gene and ORF 1ab. We describe a set of predicted spike protein epitopes whose variability is negligible. Additionally, all predicted epitopes for the structural E, M and N proteins are highly conserved. The amino acid changes present in the spike glycoprotein of variables of concern (VOCs) comprise between 3.4% and 20.7% of the predicted epitopes of this protein. These results favors the continuous efficacy of the available vaccines targeting the spike protein, and other structural proteins. Multiple epitopes vaccines should sustain vaccine efficacy since at least some of the epitopes present in variability regions of VOCs are conserved and thus recognizable by antibodies.
由严重急性呼吸综合征冠状病毒2(SARS-CoV-2)引起的2019冠状病毒病(COVID-19)大流行带来了无数挑战,比如要弄清楚是什么引发了这种新型人类病毒的出现、这种RNA病毒如何进化,或者病毒基因组的变异性如何影响作为疫苗靶点的蛋白质的一级结构。我们分析了全球流感共享数据库(GISAID)中可获取的19471个SARS-CoV-2基因组以及美国国立生物技术信息中心(NCBI)的基因银行(GenBank)中可获取的3335个其他冠状病毒科成员的基因组,收集了SARS-CoV-2高质量基因组和不同的冠状病毒科基因组。此外,我们还分析了199984个刺突糖蛋白序列。在此,我们鉴定出一个SARS-CoV-2新兴簇,其中包含13个从蝙蝠和穿山甲中分离出的密切相关基因组,这些基因组显示出重组迹象,这可能促成了SARS-CoV-2的出现。分析的SARS-CoV-2基因组呈现出9632个单核苷酸变异(SNV),对应于全基因组0.3的变异密度,且具有明显的地理分布。SNV在整个基因组中分布不均,在刺突基因和开放阅读框1ab(ORF 1ab)中发现了突变热点。我们描述了一组预测的刺突蛋白表位,其变异性可忽略不计。此外,所有预测的结构蛋白E、M和N的表位都高度保守。值得关注的变异株(VOC)的刺突糖蛋白中存在的氨基酸变化占该蛋白预测表位的3.4%至20.7%。这些结果支持了现有的针对刺突蛋白和其他结构蛋白的疫苗的持续有效性。多种表位疫苗应能维持疫苗效力,因为至少一些存在于VOC变异区域的表位是保守的,因此可被抗体识别。