Institute of Biochemistry and Biophysics, University of Tehran, Tehran, Iran.
Faculty of New Sciences and Technologies, University of Tehran, Tehran, Iran.
BMC Bioinformatics. 2021 Aug 30;22(1):416. doi: 10.1186/s12859-021-04338-7.
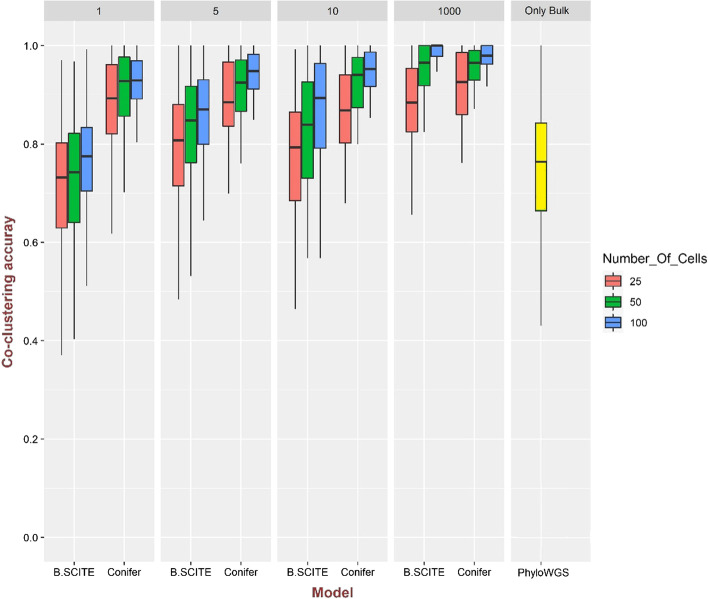
Genetic heterogeneity of a cancer tumor that develops during clonal evolution is one of the reasons for cancer treatment failure, by increasing the chance of drug resistance. Clones are cell populations with different genotypes, resulting from differences in somatic mutations that occur and accumulate during cancer development. An appropriate approach for identifying clones is determining the variant allele frequency of mutations that occurred in the tumor. Although bulk sequencing data can be used to provide that information, the frequencies are not informative enough for identifying different clones with the same prevalence and their evolutionary relationships. On the other hand, single-cell sequencing data provides valuable information about branching events in the evolution of a cancerous tumor. However, the temporal order of mutations may be determined with ambiguities using only single-cell data, while variant allele frequencies from bulk sequencing data can provide beneficial information for inferring the temporal order of mutations with fewer ambiguities.
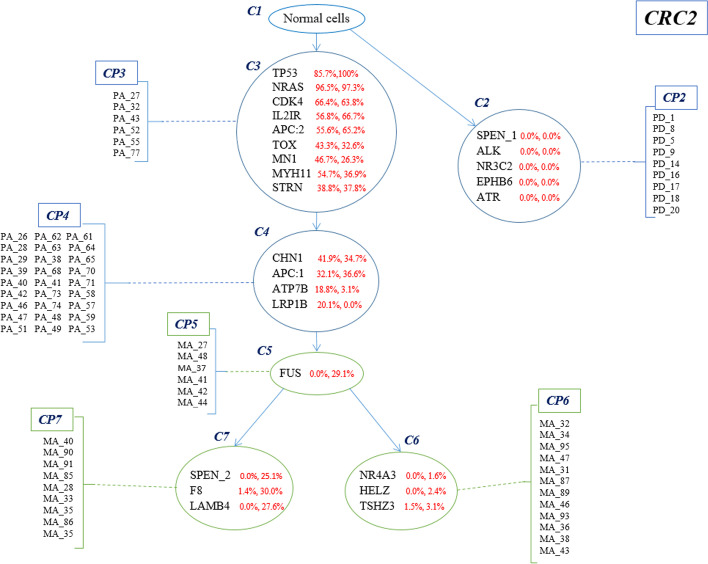
In this study, a new method called Conifer (ClONal tree Inference For hEterogeneity of tumoR) is proposed which combines aggregated variant allele frequency from bulk sequencing data with branching event information from single-cell sequencing data to more accurately identify clones and their evolutionary relationships. It is proven that the accuracy of clone identification and clonal tree inference is increased by using Conifer compared to other existing methods on various sets of simulated data. In addition, it is discussed that the evolutionary tree provided by Conifer on real cancer data sets is highly consistent with information in both bulk and single-cell data.
In this study, we have provided an accurate and robust method to identify clones of tumor heterogeneity and their evolutionary history by combining single-cell and bulk sequencing data.
在克隆进化过程中发生的肿瘤遗传异质性是癌症治疗失败的原因之一,因为它增加了产生耐药性的机会。克隆是具有不同基因型的细胞群体,是由于癌症发展过程中发生和积累的体细胞突变的差异造成的。确定克隆的一种适当方法是确定肿瘤中发生的突变的变异等位基因频率。尽管批量测序数据可用于提供该信息,但这些频率不足以识别具有相同流行率的不同克隆及其进化关系。另一方面,单细胞测序数据为癌症肿瘤进化中的分支事件提供了有价值的信息。然而,仅使用单细胞数据可能会导致突变的时间顺序存在歧义,而批量测序数据中的变异等位基因频率可以提供有用的信息,以便在较少歧义的情况下推断突变的时间顺序。
在这项研究中,提出了一种称为 Conifer(肿瘤异质性的克隆树推断)的新方法,该方法将批量测序数据的聚合变异等位基因频率与单细胞测序数据的分支事件信息结合起来,以更准确地识别克隆及其进化关系。与其他现有方法相比,在各种模拟数据集上使用 Conifer 可提高克隆识别和克隆树推断的准确性。此外,还讨论了 Conifer 在真实癌症数据集上提供的进化树与批量和单细胞数据中的信息高度一致。
在这项研究中,我们通过结合单细胞和批量测序数据,提供了一种准确而稳健的方法来识别肿瘤异质性的克隆及其进化历史。