Department of Mathematics and Program in Applied and Computational Mathematics, Princeton University, Princeton, NJ 08544, USA.
Departments of Cellular and Molecular Physiology, Biomedical Engineering, and Molecular Biophysics and Biochemistry, Yale University, New Haven, CT 06520, USA.
Annu Rev Biomed Data Sci. 2020 Jul;3:163-190. doi: 10.1146/annurev-biodatasci-021020-093826. Epub 2020 May 4.
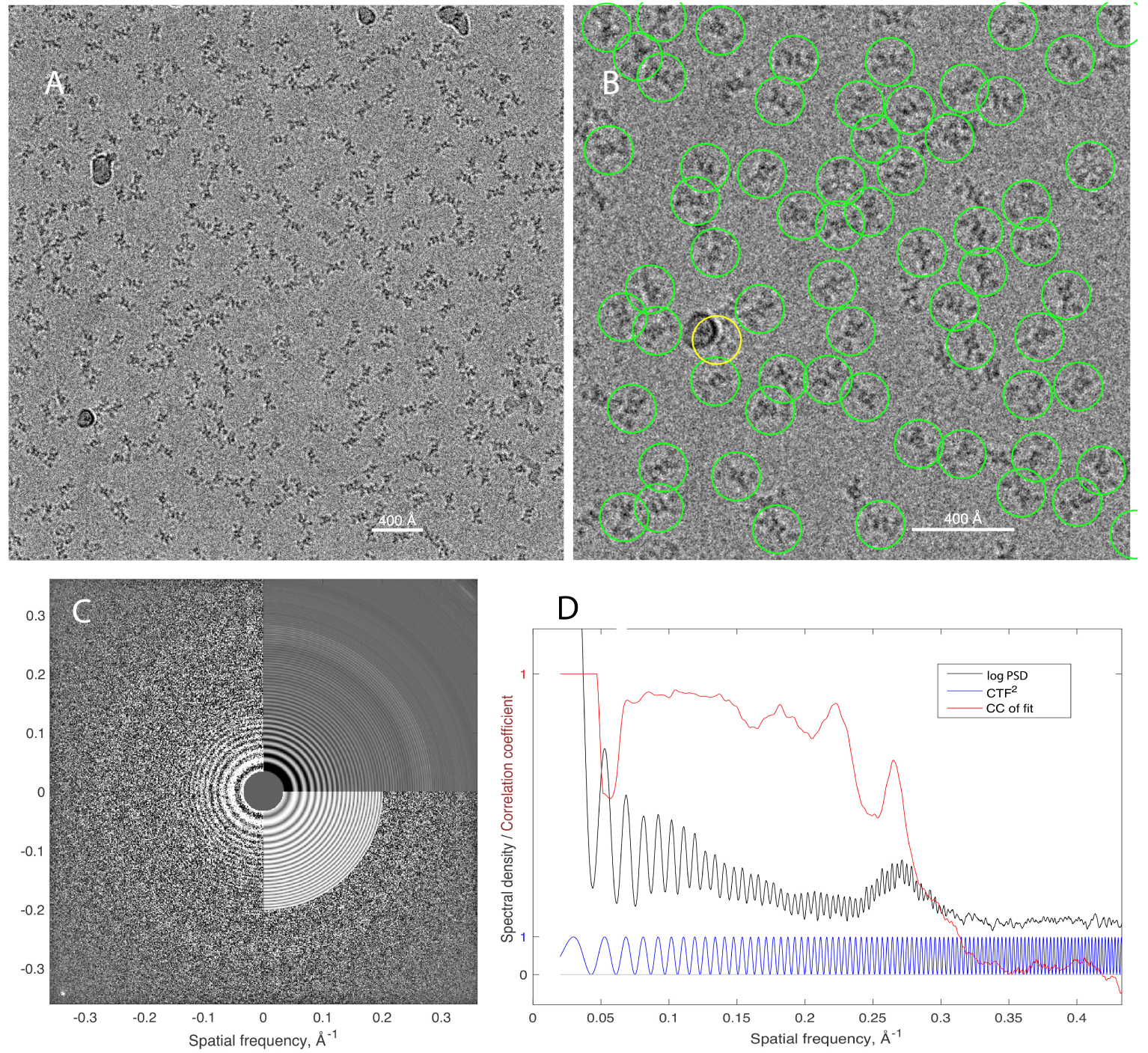
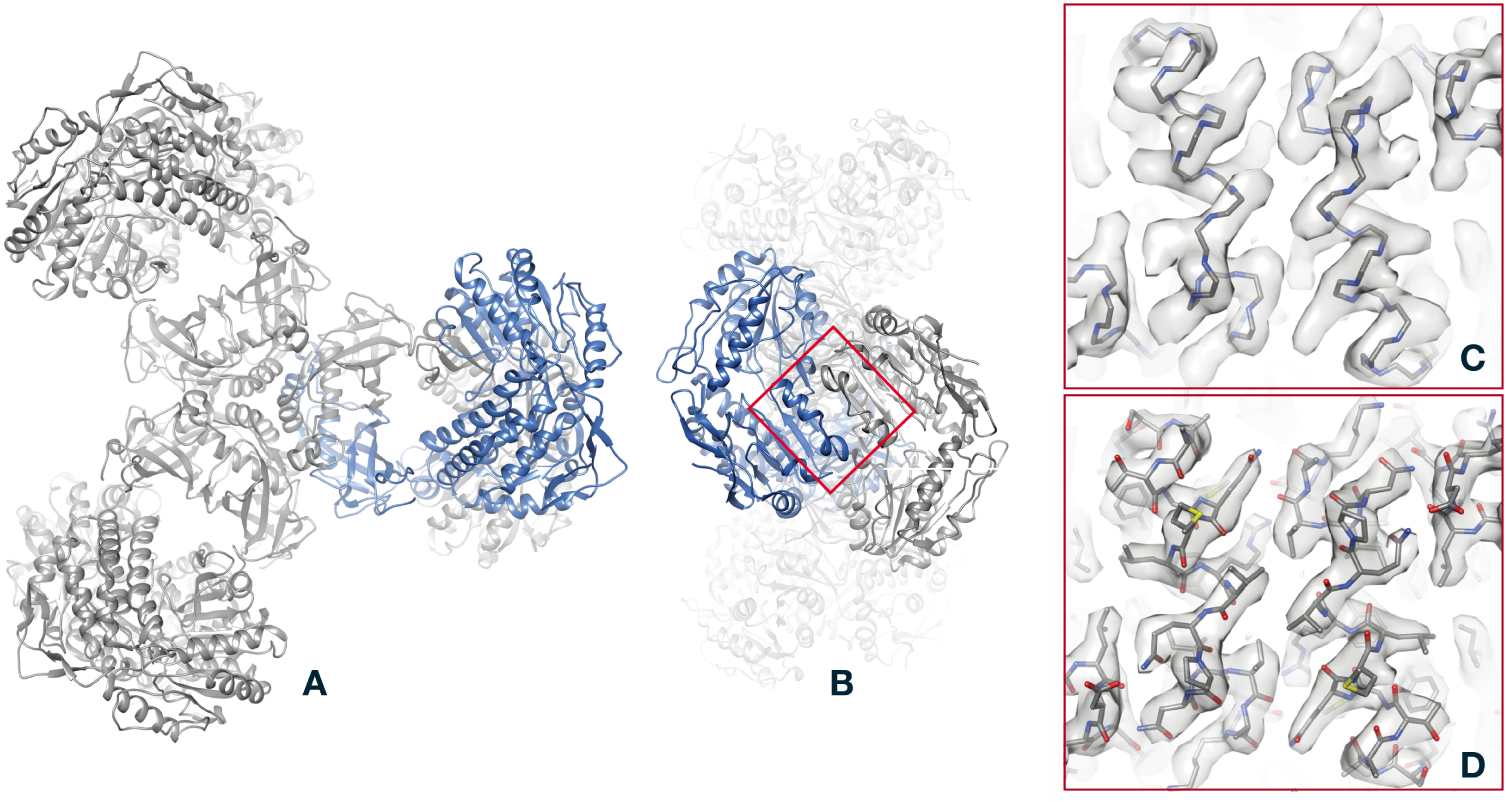
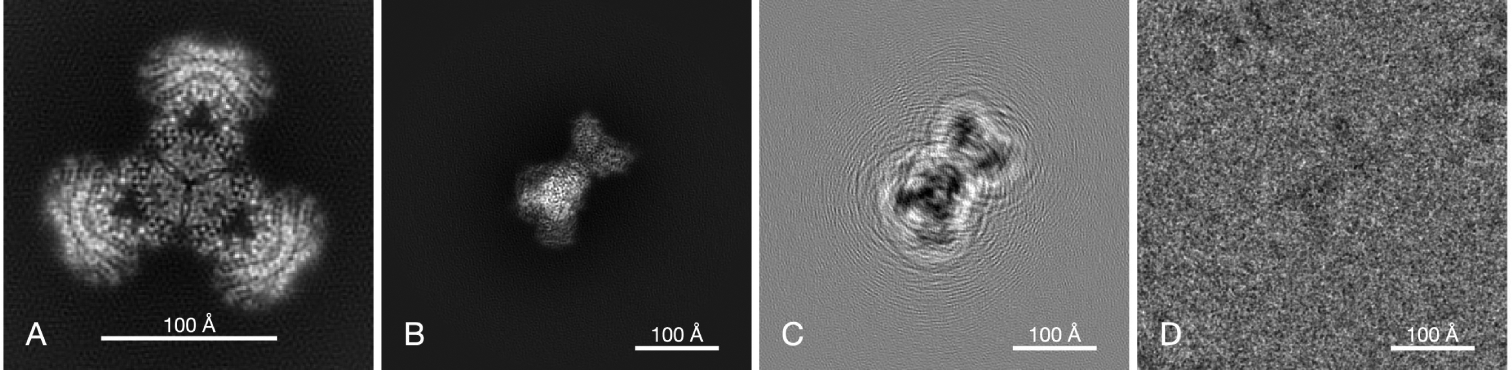
Single-particle electron cryomicroscopy (cryo-EM) is an increasingly popular technique for elucidating the three-dimensional structure of proteins and other biologically significant complexes at near-atomic resolution. It is an imaging method that does not require crystallization and can capture molecules in their native states. In single-particle cryo-EM, the three-dimensional molecular structure needs to be determined from many noisy two-dimensional tomographic projections of individual molecules, whose orientations and positions are unknown. The high level of noise and the unknown pose parameters are two key elements that make reconstruction a challenging computational problem. Even more challenging is the inference of structural variability and flexible motions when the individual molecules being imaged are in different conformational states. This review discusses computational methods for structure determination by single-particle cryo-EM and their guiding principles from statistical inference, machine learning, and signal processing that also play a significant role in many other data science applications.
单颗粒电子冷冻显微镜(cryo-EM)是一种越来越流行的技术,可用于阐明蛋白质和其他具有生物学意义的复合物的三维结构,分辨率接近原子级。它是一种不需要结晶的成像方法,可以捕获天然状态下的分子。在单颗粒 cryo-EM 中,需要从单个分子的许多嘈杂二维断层扫描投影中确定三维分子结构,而这些分子的取向和位置是未知的。高噪声水平和未知的姿势参数是使得重建成为具有挑战性的计算问题的两个关键因素。当被成像的单个分子处于不同构象状态时,推断结构变异性和柔性运动更加具有挑战性。本文综述了通过单颗粒 cryo-EM 进行结构测定的计算方法及其从统计推断、机器学习和信号处理中得出的指导原则,这些原则在许多其他数据科学应用中也起着重要作用。