Department of Gynecology, Heilongjiang University of Traditional Chinese Medicine, Harbin, PR China.
Department of Acupuncture and Moxibustion, Heilongjiang University of Traditional Chinese Medicine, Harbin, P.R. China.
World J Surg Oncol. 2021 Sep 16;19(1):277. doi: 10.1186/s12957-021-02384-2.
This study aimed to establish a risk model of hub genes to evaluate the prognosis of patients with cervical cancer.
Based on TCGA and GTEx databases, the differentially expressed genes (DEGs) were screened and then analyzed using GO and KEGG analyses. The weighted gene co-expression network (WGCNA) was then used to perform modular analysis of DEGs. Univariate Cox regression analysis combined with LASSO and Cox-pH was used to select the prognostic genes. Then, multivariate Cox regression analysis was used to screen the hub genes. The risk model was established based on hub genes and evaluated by risk curve, survival state, Kaplan-Meier curve, and receiver operating characteristic (ROC) curve.
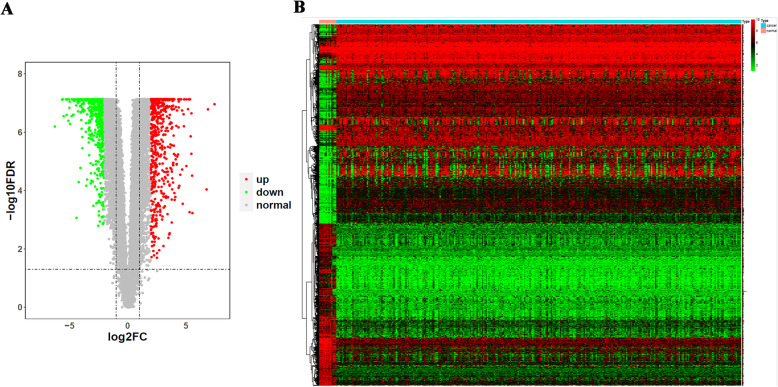
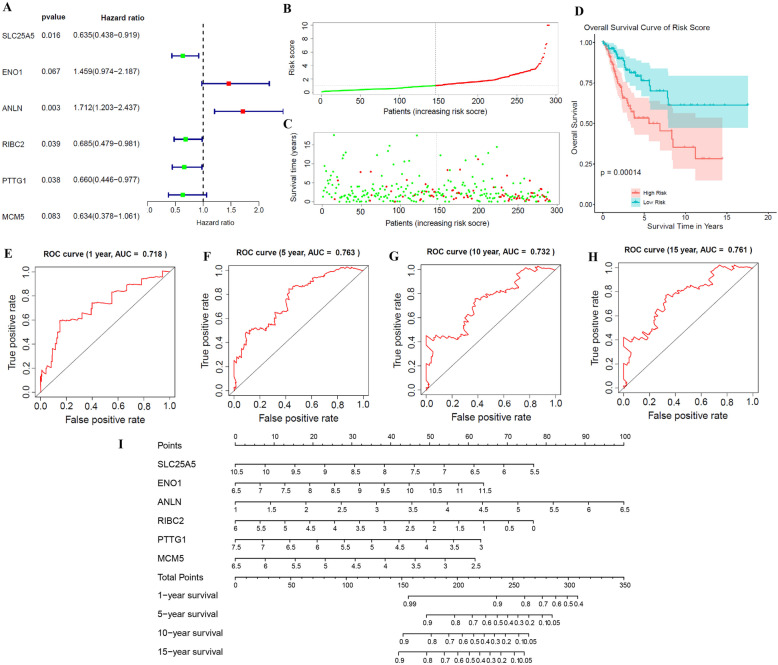
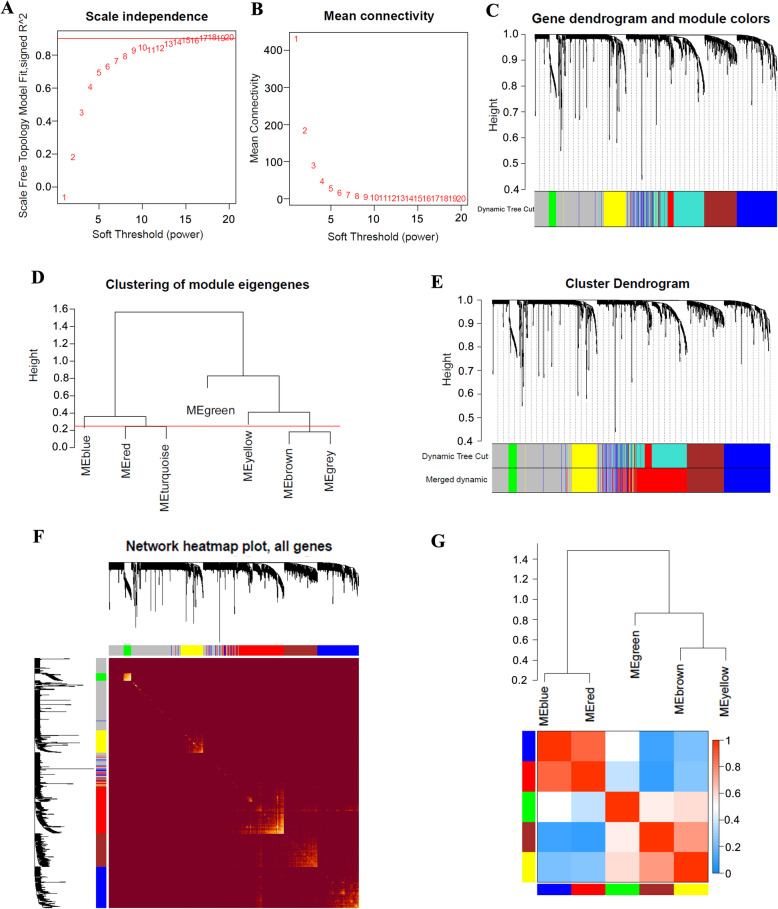
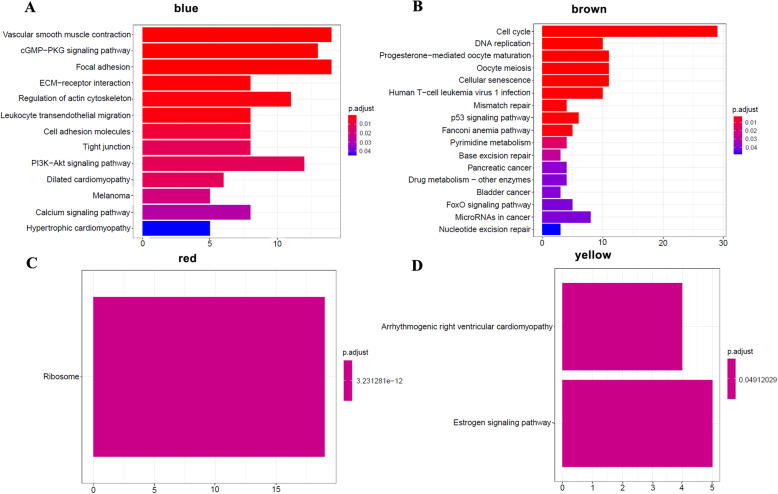
We screened 1265 DEGs between cervical cancer and normal samples, of which 620 were downregulated and 645 were upregulated. GO and KEGG analyses revealed that most of the upregulated genes were related to the metastasis of cancer cells, while the downregulated genes mostly acted on the cell cycle. Then, WGCNA mined six modules (red, blue, green, brown, yellow, and gray), and the brown module with the most DEGs and related to multiple cancers was selected for the follow-up study. Eight genes were identified by univariate Cox regression analysis combined with the LASSO Cox-pH model. Then, six hub genes (SLC25A5, ENO1, ANLN, RIBC2, PTTG1, and MCM5) were screened by multivariate Cox regression analysis, and SLC25A5, ANLN, RIBC2, and PTTG1 could be used as independent prognostic factors. Finally, we determined that the risk model established by the six hub genes was effective and stable.
This study supplies the prognostic value of the risk model and the new promising targets for the cervical cancer treatment, and their biological functions need to be further explored.
本研究旨在建立一个评估宫颈癌患者预后的关键基因风险模型。
基于 TCGA 和 GTEx 数据库,筛选差异表达基因(DEGs),并进行 GO 和 KEGG 分析。然后,使用加权基因共表达网络(WGCNA)对 DEGs 进行模块分析。采用单因素 Cox 回归分析联合 LASSO 和 Cox-PH 筛选预后基因。然后,采用多因素 Cox 回归分析筛选关键基因。基于关键基因建立风险模型,并通过风险曲线、生存状态、Kaplan-Meier 曲线和受试者工作特征(ROC)曲线进行评估。
我们筛选出宫颈癌与正常样本之间的 1265 个 DEGs,其中 620 个下调,645 个上调。GO 和 KEGG 分析表明,大多数上调基因与癌细胞的转移有关,而下调基因主要作用于细胞周期。然后,WGCNA 挖掘出六个模块(红色、蓝色、绿色、棕色、黄色和灰色),并选择与多种癌症相关且具有最多 DEGs 的棕色模块进行后续研究。通过单因素 Cox 回归分析联合 LASSO Cox-PH 模型,识别出 8 个基因。然后,通过多因素 Cox 回归分析筛选出 6 个关键基因(SLC25A5、ENO1、ANLN、RIBC2、PTTG1 和 MCM5),其中 SLC25A5、ANLN、RIBC2 和 PTTG1 可作为独立的预后因素。最后,我们确定由六个关键基因建立的风险模型是有效的和稳定的。
本研究为宫颈癌的治疗提供了预后价值和新的有前途的靶点,其生物学功能有待进一步探索。