Sicho M, Liu X, Svozil D, van Westen G J P
CZ-OPENSCREEN: National Infrastructure for Chemical Biology, Department of Informatics and Chemistry, Faculty of Chemical Technology, University of Chemistry and Technology Prague, Technická 5, 166 28, Prague, Czech Republic.
Computational Drug Discovery, Drug Discovery and Safety, Leiden Academic Centre for Drug Research, Einsteinweg 55, Leiden, The Netherlands.
J Cheminform. 2021 Sep 25;13(1):73. doi: 10.1186/s13321-021-00550-y.
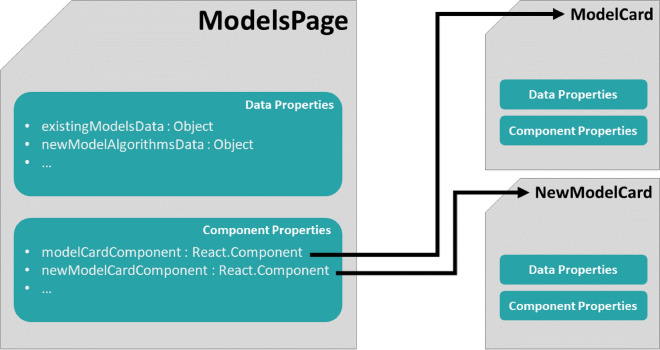
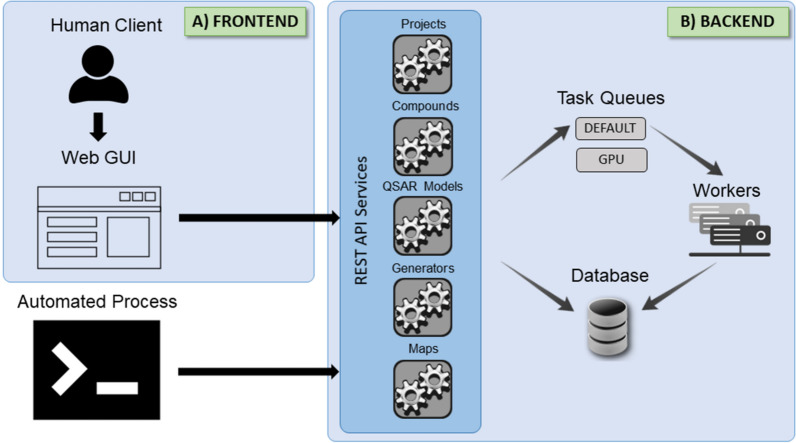
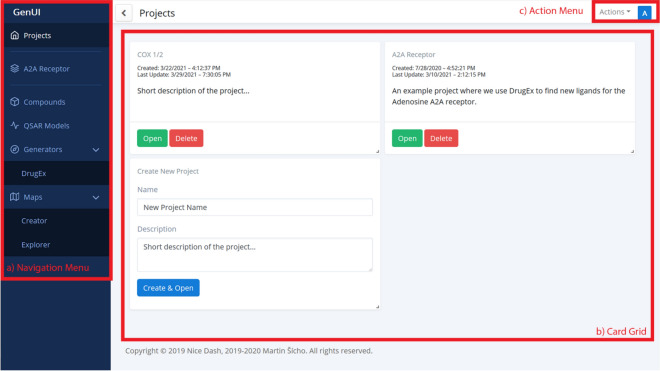
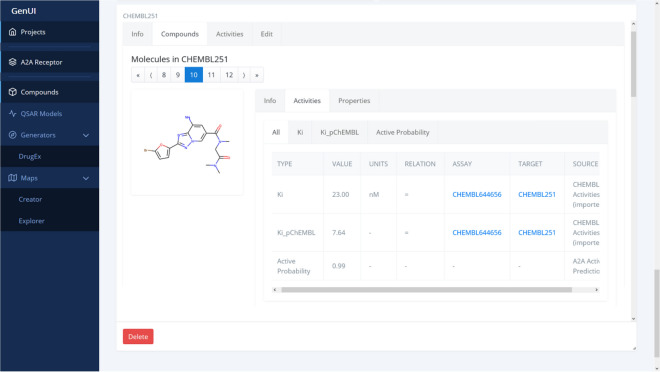
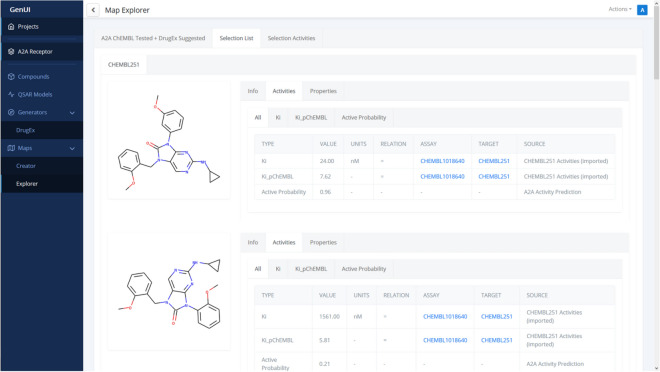
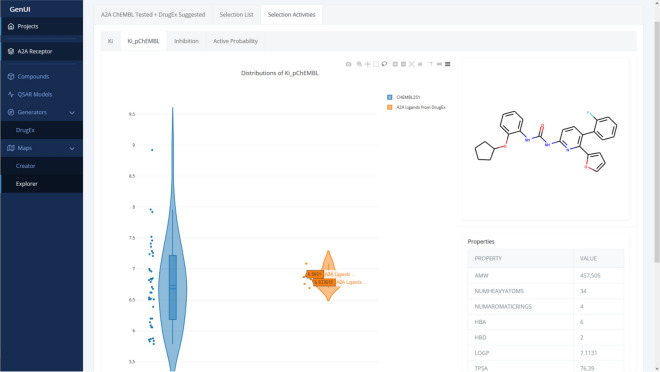
Many contemporary cheminformatics methods, including computer-aided de novo drug design, hold promise to significantly accelerate and reduce the cost of drug discovery. Thanks to this attractive outlook, the field has thrived and in the past few years has seen an especially significant growth, mainly due to the emergence of novel methods based on deep neural networks. This growth is also apparent in the development of novel de novo drug design methods with many new generative algorithms now available. However, widespread adoption of new generative techniques in the fields like medicinal chemistry or chemical biology is still lagging behind the most recent developments. Upon taking a closer look, this fact is not surprising since in order to successfully integrate the most recent de novo drug design methods in existing processes and pipelines, a close collaboration between diverse groups of experimental and theoretical scientists needs to be established. Therefore, to accelerate the adoption of both modern and traditional de novo molecular generators, we developed Generator User Interface (GenUI), a software platform that makes it possible to integrate molecular generators within a feature-rich graphical user interface that is easy to use by experts of diverse backgrounds. GenUI is implemented as a web service and its interfaces offer access to cheminformatics tools for data preprocessing, model building, molecule generation, and interactive chemical space visualization. Moreover, the platform is easy to extend with customizable frontend React.js components and backend Python extensions. GenUI is open source and a recently developed de novo molecular generator, DrugEx, was integrated as a proof of principle. In this work, we present the architecture and implementation details of GenUI and discuss how it can facilitate collaboration in the disparate communities interested in de novo molecular generation and computer-aided drug discovery.
许多当代化学信息学方法,包括计算机辅助从头药物设计,有望显著加速并降低药物发现的成本。得益于这一诱人的前景,该领域蓬勃发展,在过去几年中尤其取得了显著增长,这主要归功于基于深度神经网络的新方法的出现。这种增长在新型从头药物设计方法的发展中也很明显,现在有许多新的生成算法可用。然而,新的生成技术在药物化学或化学生物学等领域的广泛应用仍落后于最新发展。仔细观察就会发现,这一事实并不奇怪,因为为了将最新的从头药物设计方法成功整合到现有流程和管道中,需要在不同的实验科学家和理论科学家群体之间建立密切合作。因此,为了加速现代和传统从头分子生成器的采用,我们开发了生成器用户界面(GenUI),这是一个软件平台,它能够在一个功能丰富的图形用户界面中集成分子生成器,便于不同背景的专家使用。GenUI作为一个网络服务来实现,其接口提供对化学信息学工具的访问,用于数据预处理、模型构建、分子生成和交互式化学空间可视化。此外,该平台很容易通过可定制的前端React.js组件和后端Python扩展进行扩展。GenUI是开源的,最近开发的一种从头分子生成器DrugEx已作为原理验证被集成进来。在这项工作中,我们展示了GenUI的架构和实现细节,并讨论了它如何促进对从头分子生成和计算机辅助药物发现感兴趣的不同群体之间的合作。