Allam Omar, Cho Byung Woo, Kim Ki Chul, Jang Seung Soon
Computational NanoBio Technology Laboratory, School of Materials Science and Engineering, Georgia Institute of Technology Atlanta GA 30332-0245 USA
The George W. Woodruff School of Mechanical Engineering, Georgia Institute of Technology Atlanta GA 30332-0405 USA.
RSC Adv. 2018 Nov 26;8(69):39414-39420. doi: 10.1039/c8ra07112h. eCollection 2018 Nov 23.
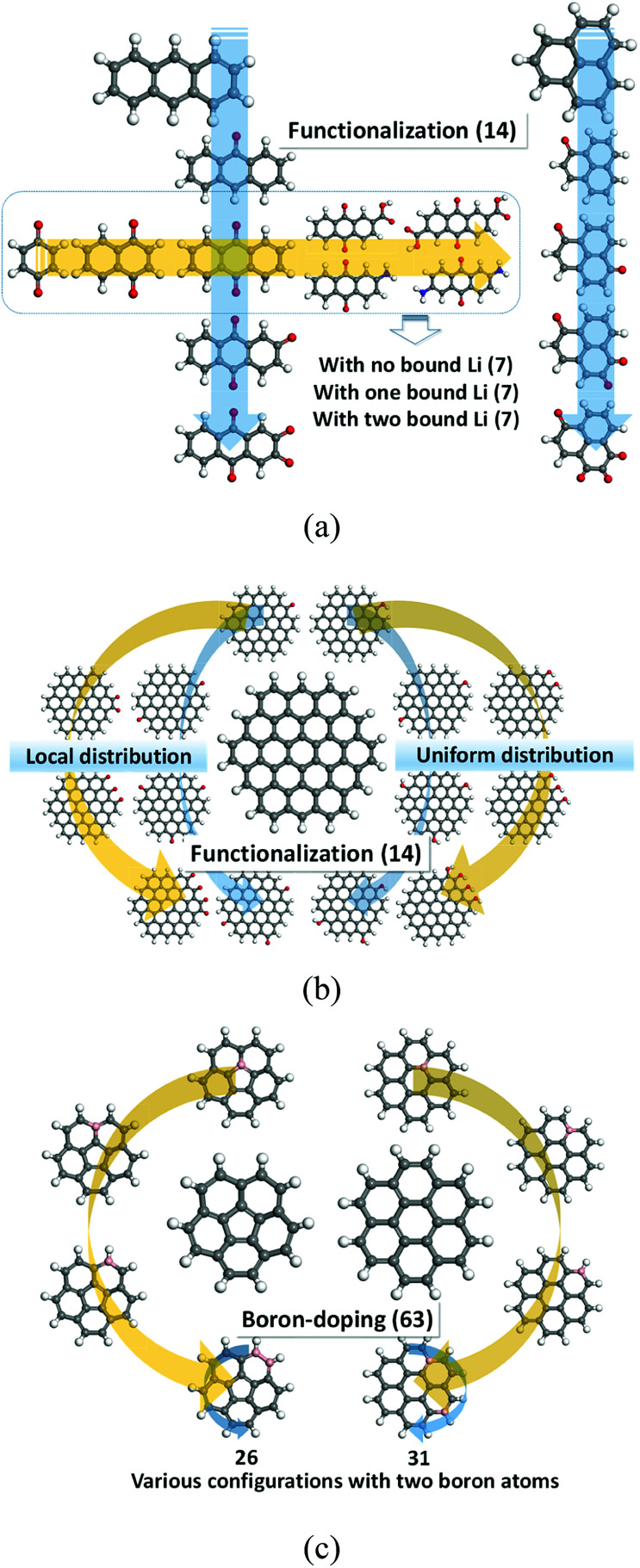
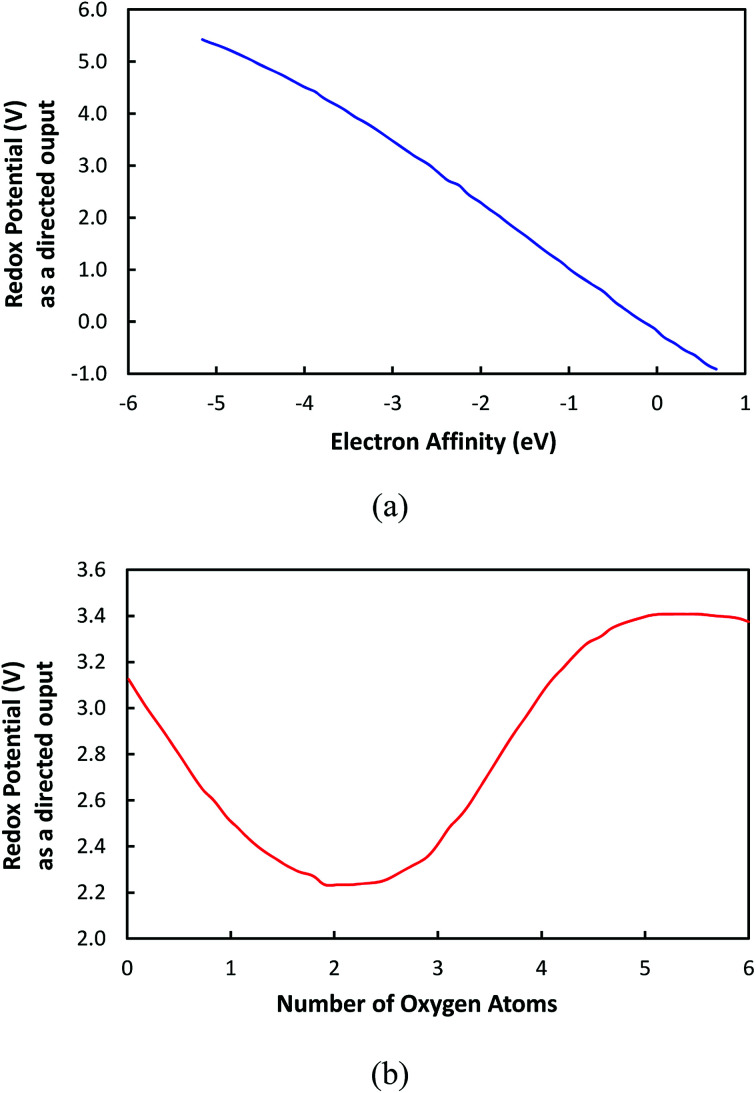
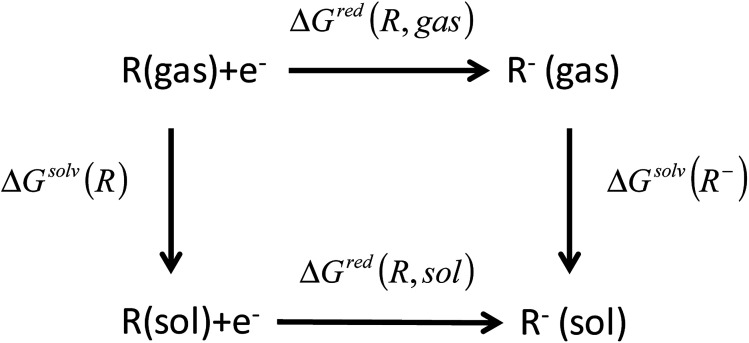
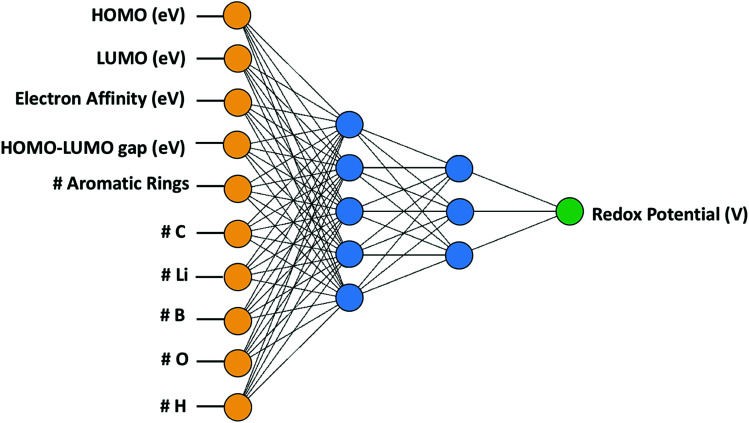
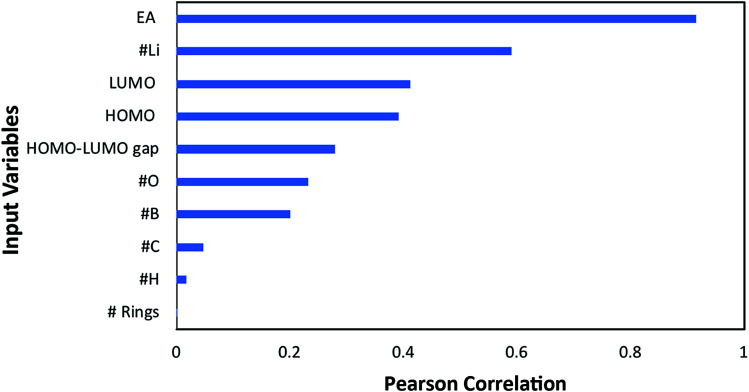
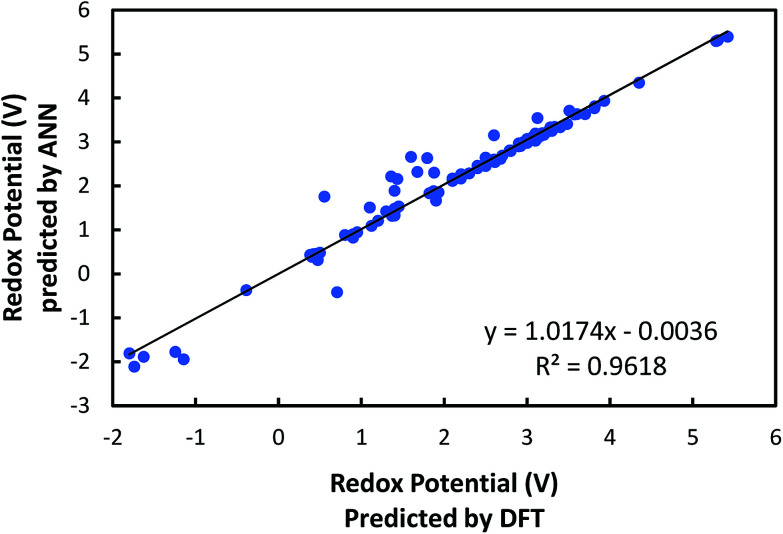
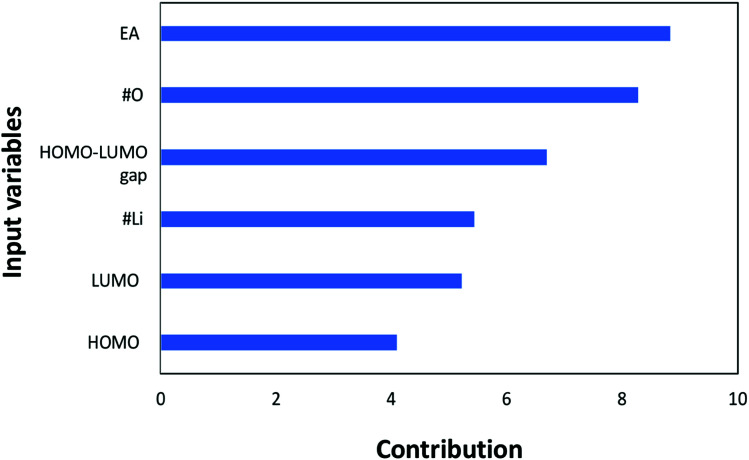
In this study, we utilize a density functional theory-machine learning framework to develop a high-throughput screening method for designing new molecular electrode materials. For this purpose, a density functional theory modeling approach is employed to predict basic quantum mechanical quantities such as redox potentials, and electronic properties such as electron affinity, highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO), for a selected set of organic materials. Both the electronic properties and structural information, such as the numbers of oxygen atoms, lithium atoms, boron atoms, carbon atoms, hydrogen atoms, and aromatic rings, are considered as input variables for the machine learning-based prediction of redox potentials. The large-set of input variables are further downsized using a linear correlation analysis to have six core input variables, namely electron affinity, HOMO, LUMO, HOMO-LUMO gap, the number of oxygen atoms and the number of lithium atoms. The artificial neural network trained using the quasi-Newton method demonstrates a capability for accurately estimating the redox potentials. From the contribution analysis, in which the influence of each input on the target are accessed, we highlight that the electron affinity has the highest contribution to redox potential, followed by the number of oxygen atoms, HOMO-LUMO gap, the number of lithium atoms, LUMO, and HOMO, in order.
在本研究中,我们利用密度泛函理论-机器学习框架开发了一种用于设计新型分子电极材料的高通量筛选方法。为此,采用密度泛函理论建模方法来预测一组选定有机材料的基本量子力学量,如氧化还原电位,以及电子性质,如电子亲和能、最高占据分子轨道(HOMO)和最低未占据分子轨道(LUMO)。电子性质和结构信息,如氧原子、锂原子、硼原子、碳原子、氢原子的数量以及芳香环的数量,都被视为基于机器学习预测氧化还原电位的输入变量。使用线性相关分析进一步精简大量输入变量,得到六个核心输入变量,即电子亲和能、HOMO、LUMO、HOMO-LUMO能隙、氧原子数量和锂原子数量。使用拟牛顿法训练的人工神经网络展示了准确估计氧化还原电位的能力。通过贡献分析(其中评估每个输入对目标的影响),我们强调电子亲和能对氧化还原电位的贡献最大,其次依次是氧原子数量、HOMO-LUMO能隙、锂原子数量、LUMO和HOMO。