Population Health Sciences Institute, Newcastle University, Newcastle upon Tyne, United Kingdom.
Translational and Clinical Research Institute, Newcastle University, Newcastle upon Tyne, United Kingdom.
PLoS Genet. 2021 Sep 29;17(9):e1009811. doi: 10.1371/journal.pgen.1009811. eCollection 2021 Sep.
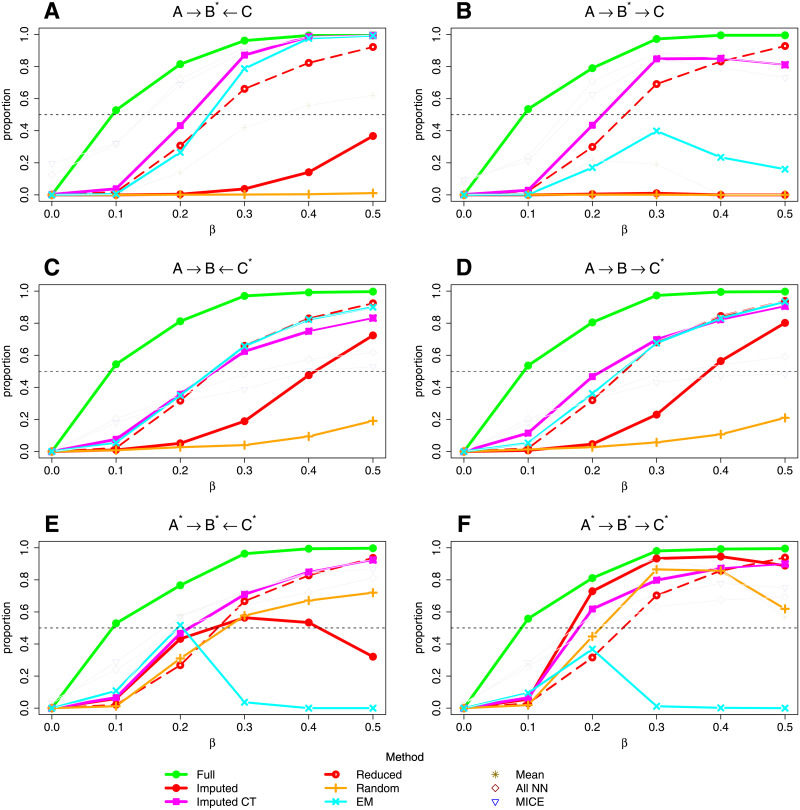
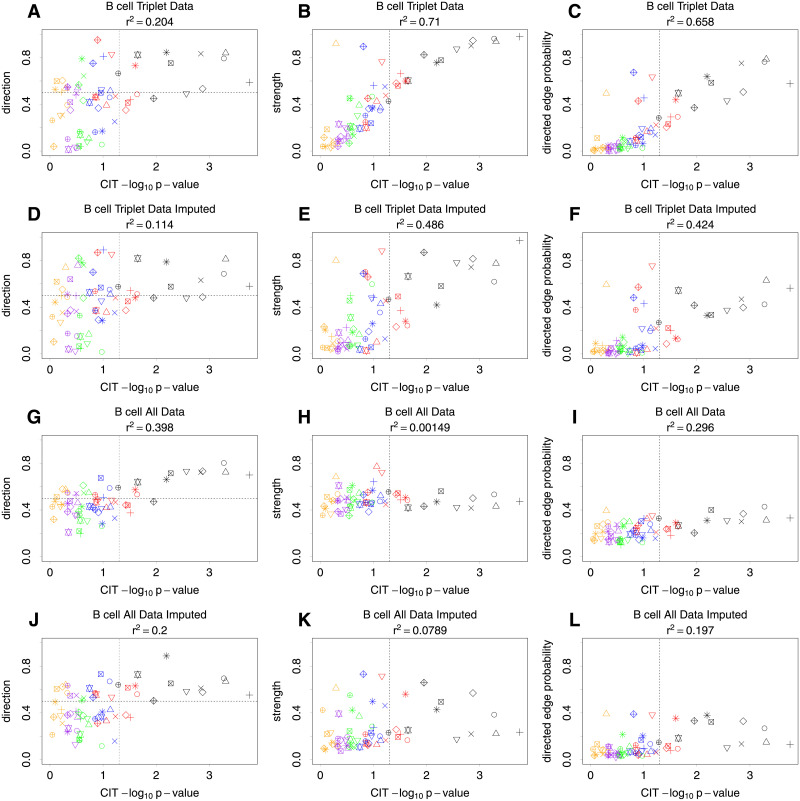
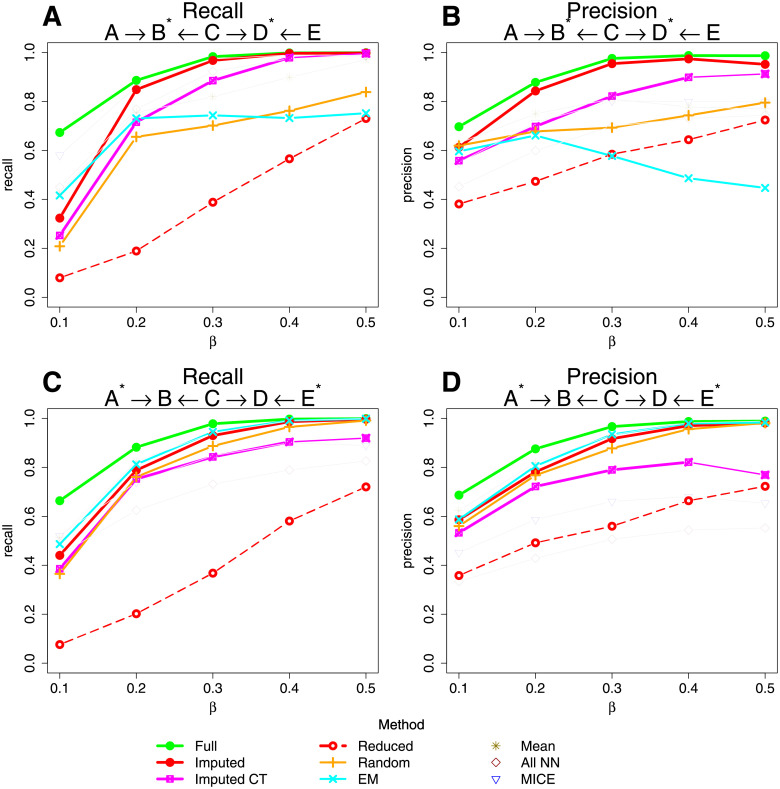
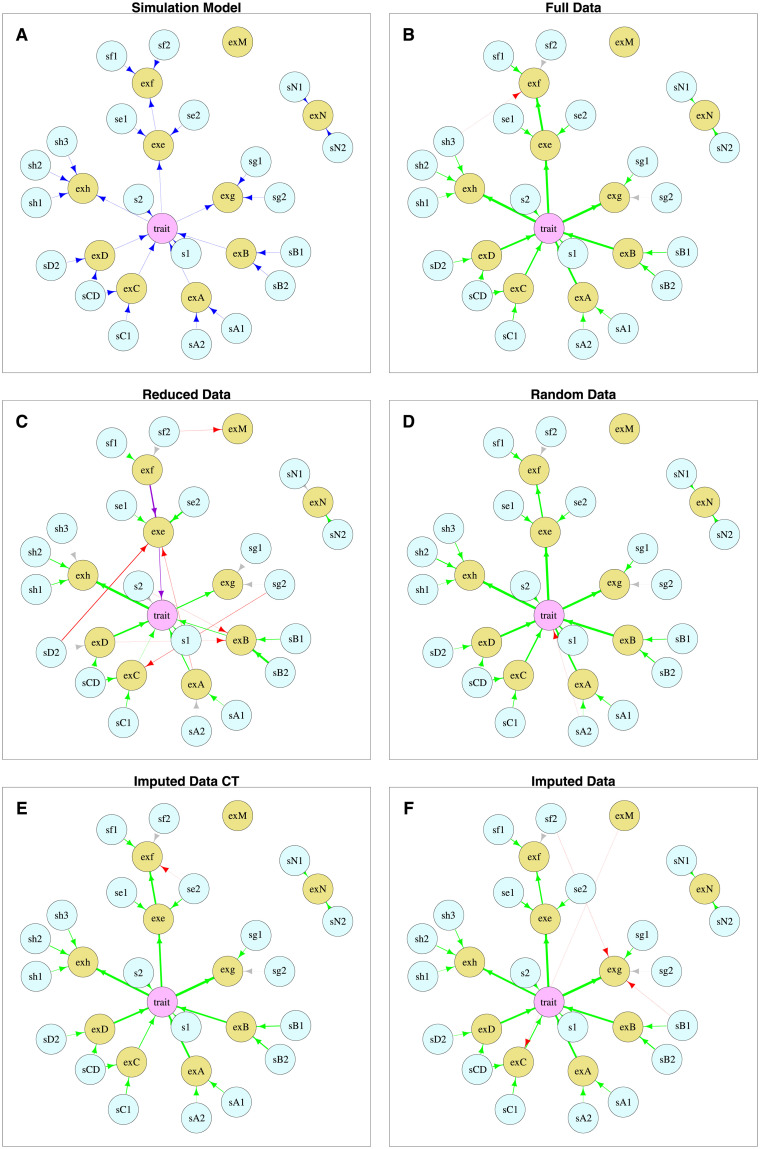
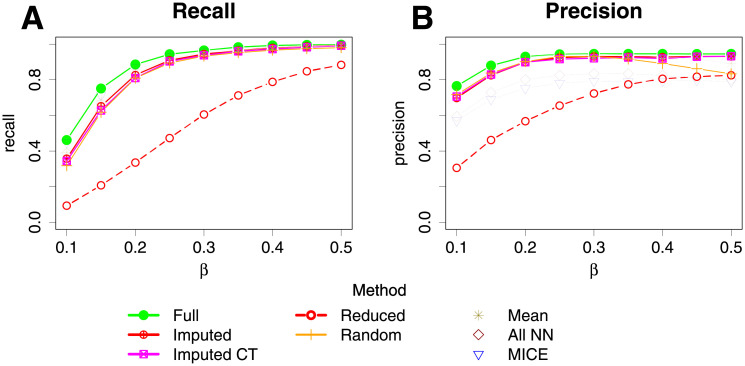
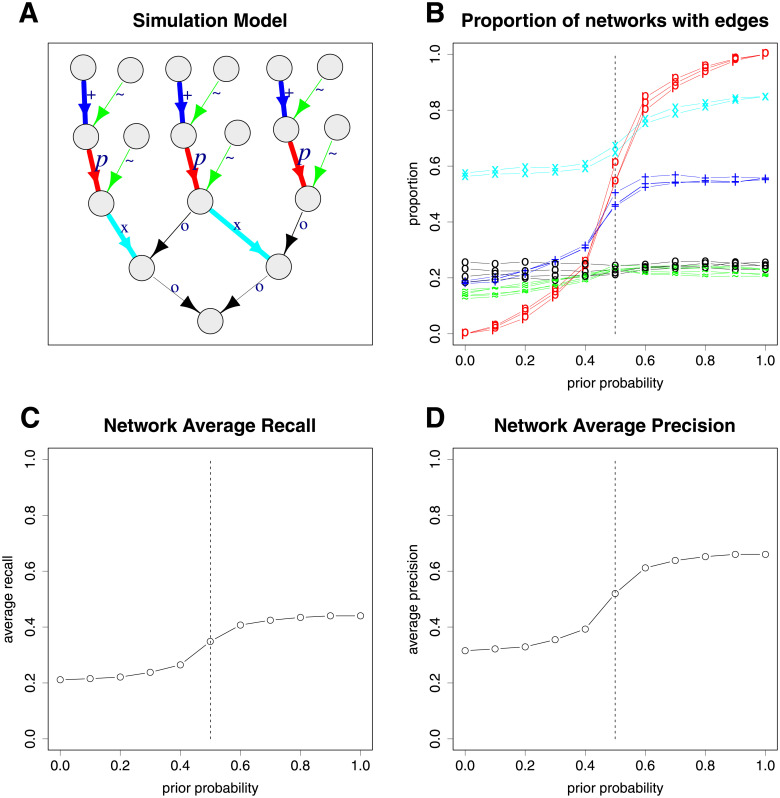
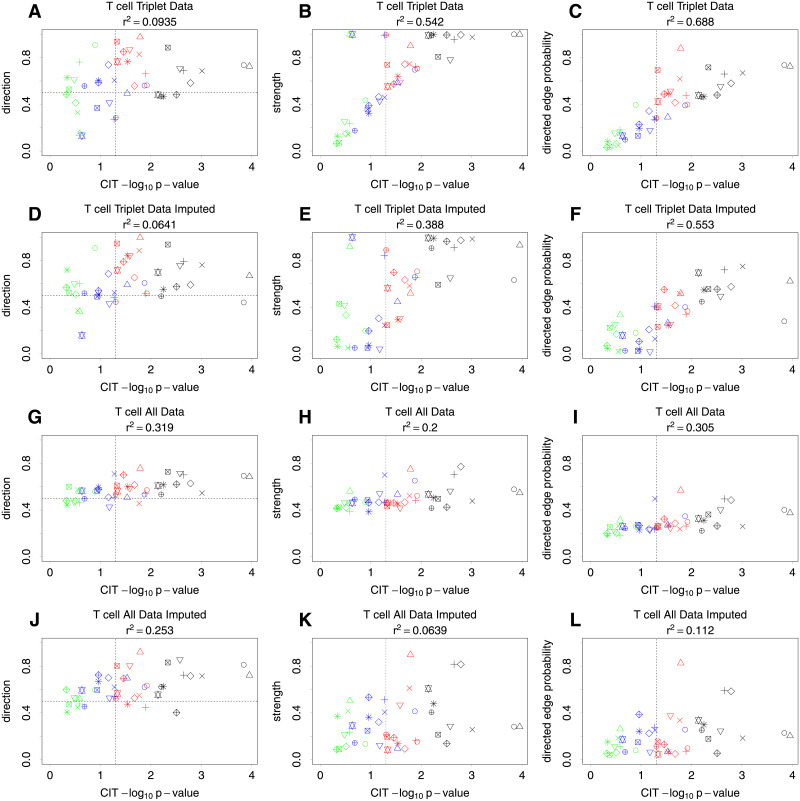
Bayesian networks can be used to identify possible causal relationships between variables based on their conditional dependencies and independencies, which can be particularly useful in complex biological scenarios with many measured variables. Here we propose two improvements to an existing method for Bayesian network analysis, designed to increase the power to detect potential causal relationships between variables (including potentially a mixture of both discrete and continuous variables). Our first improvement relates to the treatment of missing data. When there is missing data, the standard approach is to remove every individual with any missing data before performing analysis. This can be wasteful and undesirable when there are many individuals with missing data, perhaps with only one or a few variables missing. This motivates the use of imputation. We present a new imputation method that uses a version of nearest neighbour imputation, whereby missing data from one individual is replaced with data from another individual, their nearest neighbour. For each individual with missing data, the subsets of variables to be used to select the nearest neighbour are chosen by sampling without replacement the complete data and estimating a best fit Bayesian network. We show that this approach leads to marked improvements in the recall and precision of directed edges in the final network identified, and we illustrate the approach through application to data from a recent study investigating the causal relationship between methylation and gene expression in early inflammatory arthritis patients. We also describe a second improvement in the form of a pseudo-Bayesian approach for upweighting certain network edges, which can be useful when there is prior evidence concerning their directions.
贝叶斯网络可用于根据变量的条件依赖和独立性,识别变量之间可能存在的因果关系。这在具有许多测量变量的复杂生物学场景中特别有用。在这里,我们提出了对现有贝叶斯网络分析方法的两项改进,旨在提高检测变量之间潜在因果关系的能力(包括离散和连续变量的混合)。我们的第一项改进涉及缺失数据的处理。当存在缺失数据时,标准方法是在进行分析之前,删除每个存在缺失数据的个体。当存在许多缺失数据的个体时,这可能是浪费且不理想的,也许只有一个或几个变量缺失。这促使我们使用插补法。我们提出了一种新的插补方法,该方法使用了一种最近邻插补的版本,即通过用另一个个体的数据替换一个个体的缺失数据,该个体是他们的最近邻。对于每个存在缺失数据的个体,选择最近邻的变量子集是通过无放回抽样完整数据并估计最佳拟合贝叶斯网络来完成的。我们表明,这种方法可显著提高最终网络中定向边的召回率和准确率,并通过应用于最近一项研究的数据来说明该方法,该研究旨在调查早期炎症性关节炎患者中甲基化和基因表达之间的因果关系。我们还描述了第二种改进方法,即伪贝叶斯方法,用于对某些网络边缘进行加权处理,当存在有关其方向的先验证据时,这种方法很有用。