Umar Haruna Isiyaku, Ajayi Adeola, Bello Ridwan Opeyemi, Alabere Hafsat Olateju, Sanusi Afees Akinbode, Awolaja Olamide Olusegun, Alshehri Mohammed Mansour, Chukwuemeka Prosper Obed
Department of Biochemistry, School of Life Sciences, Federal University of Technology, Along Owo-Ilesha Express way, P. M. B. 704, Akure, Ondo State Nigeria.
Computer-Aided Therapeutic Discovery and Design Group, FUTA, Akure, Nigeria.
Chem Zvesti. 2022;76(2):785-796. doi: 10.1007/s11696-021-01899-y. Epub 2021 Oct 5.
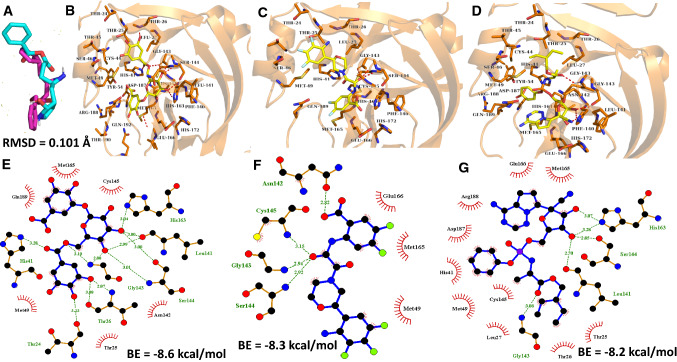
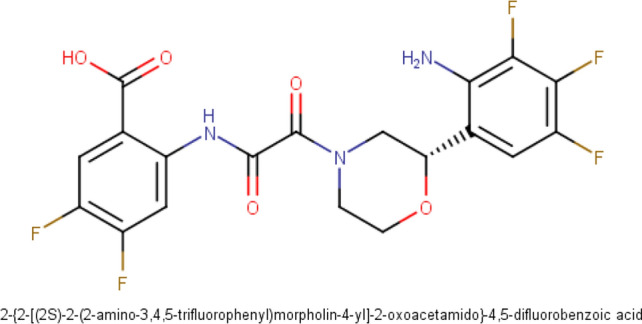
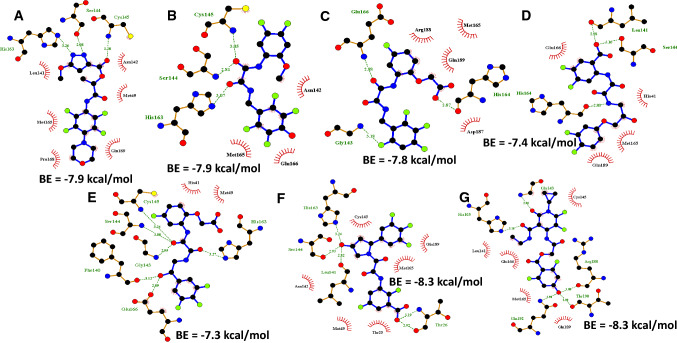
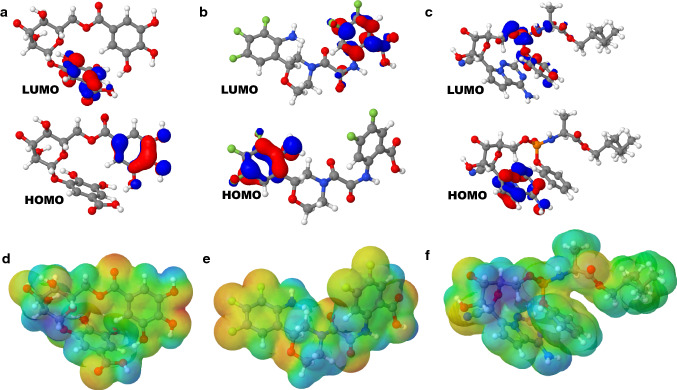
The ongoing pandemic caused by the severe acute respiratory syndrome 2 (SARS-CoV 2) has led to more than 168 million confirmed cases with 3.5 million deaths as at 28th May, 2021 across 218 countries. The virus has a cysteine protease called main protease (Mpro) which is significant to it life cycle, tagged as a suitable target for novel antivirals. In this computer-assisted study, we designed 100 novel molecules through an artificial neural network-driven platform called LigDream (https://playmolecule.org/LigDream/) using 3-O-(6-galloylglucoside) as parent molecule for design. Druglikeness screening of the molecules through five (5) different rules was carried out, followed by a virtual screening of those molecules without a single violation of the druglike rules using AutoDock Vina against Mpro. The in silico pharmacokinetic features were predicted and finally, quantum mechanics/molecular mechanics (QM/MM) study was carried out using Molecular Orbital Package 2016 (MOPAC2016) on the overall hit compound with controls to determine the stability and reactivity of the lead molecule. The findings showed that eight (8) novel molecules violated none of the druglikeness rules of which three (3) novel molecules (C33, C35 and C54) showed the utmost binding affinity of -8.3 kcal/mol against Mpro; C33 showed a good in silico pharmacokinetic features with acceptable level of stability and reactively better than our controls based on the quantum chemical descriptors analysis. However, there is an urgent need to carry out more research on these novel molecules for the fight against the disease.
The online version contains supplementary material available at 10.1007/s11696-021-01899-y.
截至2021年5月28日,由严重急性呼吸综合征冠状病毒2(SARS-CoV-2)引发的持续大流行已导致全球218个国家出现超过1.68亿确诊病例,350万人死亡。该病毒有一种名为主要蛋白酶(Mpro)的半胱氨酸蛋白酶,对其生命周期至关重要,被视为新型抗病毒药物的合适靶点。在这项计算机辅助研究中,我们通过一个名为LigDream(https://playmolecule.org/LigDream/)的人工神经网络驱动平台,以3-O-(6-没食子酰葡萄糖苷)为母体分子设计了100种新型分子。通过五条不同规则对这些分子进行类药性质筛选,然后使用AutoDock Vina对未违反任何类药规则的分子针对Mpro进行虚拟筛选。预测了计算机模拟的药代动力学特征,最后,使用分子轨道软件包2016(MOPAC2016)对整体命中化合物及对照进行量子力学/分子力学(QM/MM)研究,以确定先导分子的稳定性和反应活性。研究结果表明,有八种新型分子未违反任何类药规则,其中三种新型分子(C33、C35和C54)对Mpro表现出最高的结合亲和力,为-8.3千卡/摩尔;根据量子化学描述符分析,C33显示出良好的计算机模拟药代动力学特征,稳定性可接受,反应活性优于我们的对照。然而,迫切需要对这些新型分子进行更多研究以对抗该疾病。
在线版本包含可在10.1007/s11696-021-01899-y获取的补充材料。