Daoui Ossama, Elkhattabi Souad, Chtita Samir
Laboratory of Engineering, Systems and Applications, National School of Applied Sciences, Sidi Mohamed Ben Abdellah-Fez University, BP Box 72, Fez, Morocco.
Laboratory of Analytical and Molecular Chemistry, Faculty of Sciences Ben M'Sik, Hassan II University of Casablanca, B.P 7955 Casablanca, Morocco.
Struct Chem. 2022;33(5):1667-1690. doi: 10.1007/s11224-022-02004-z. Epub 2022 Jul 7.
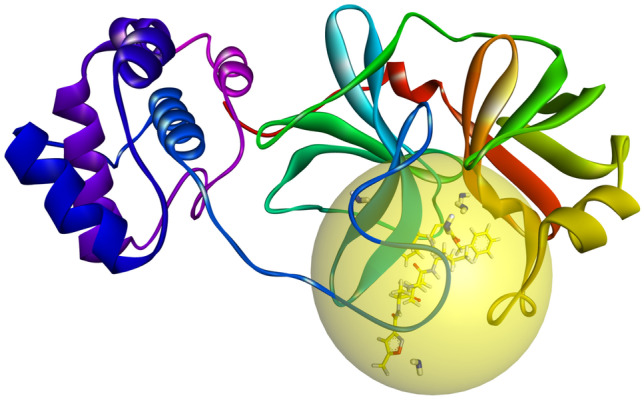
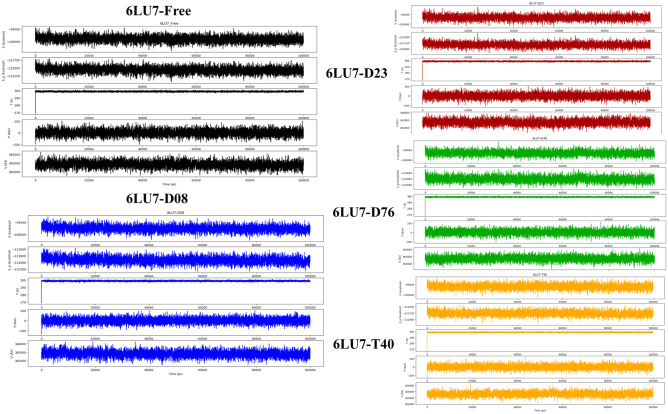
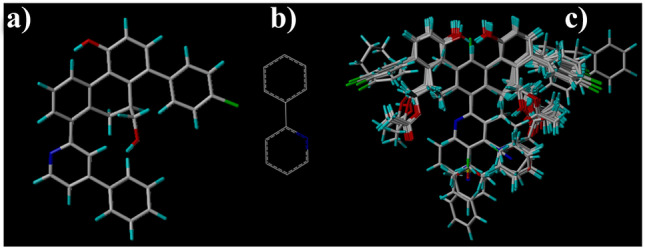
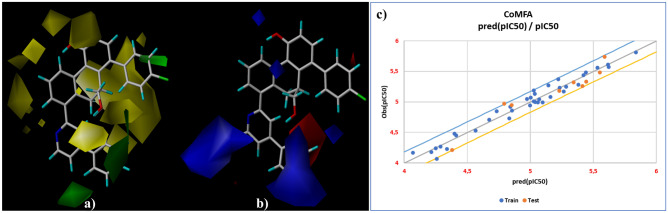
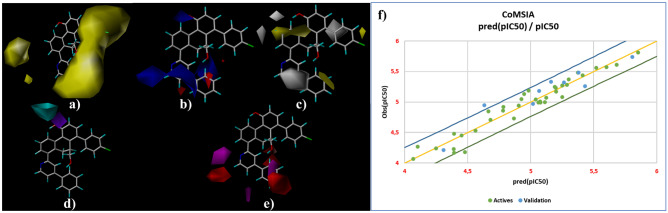
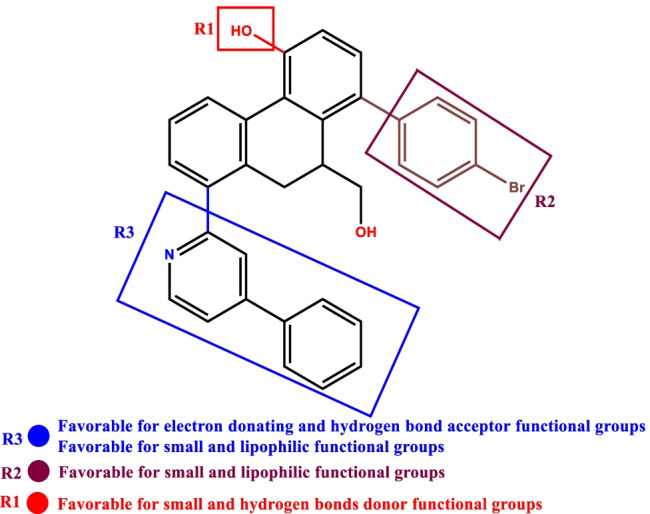
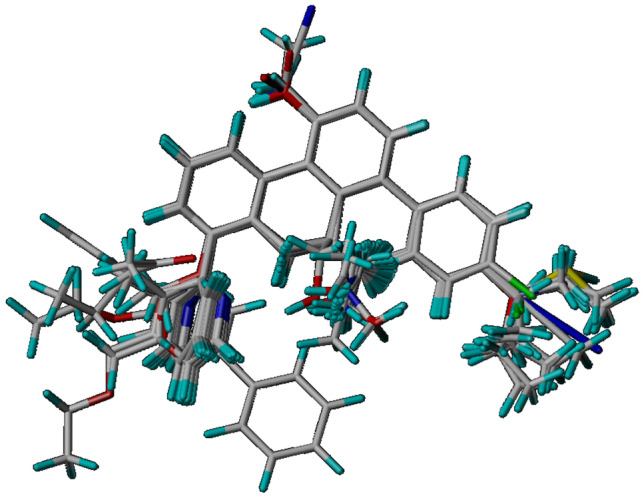
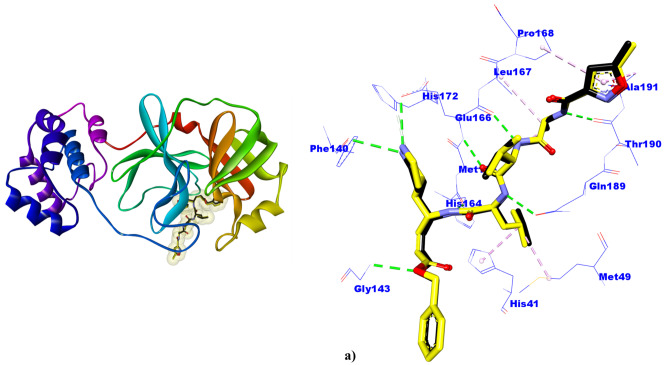
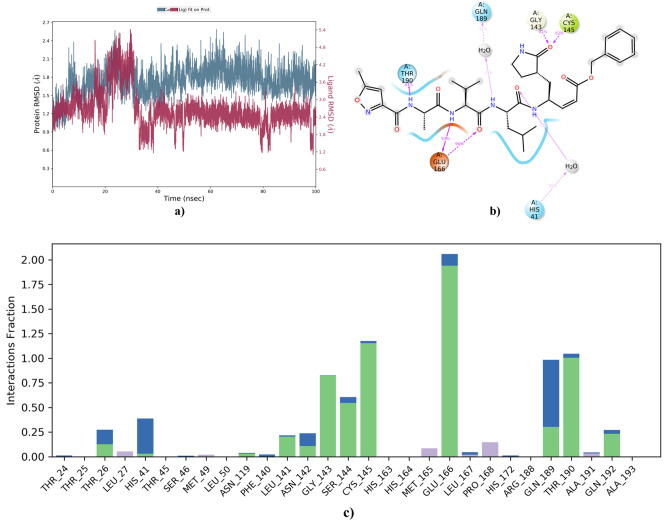
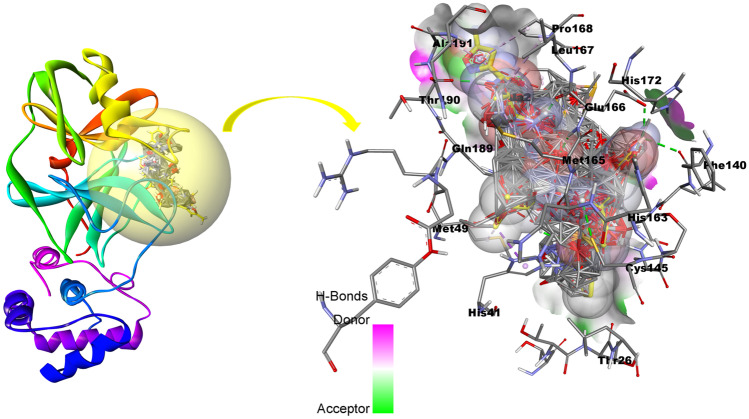
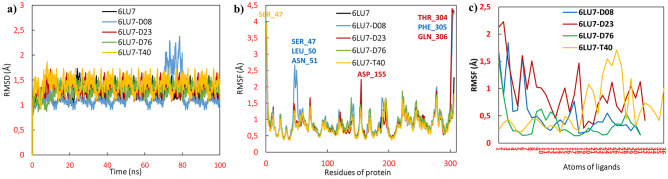
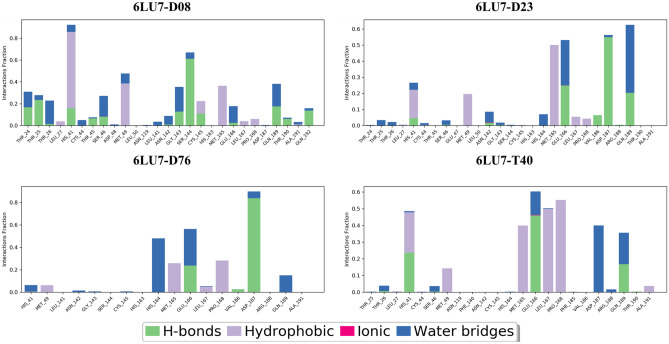
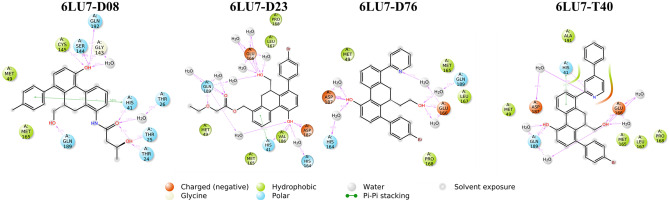
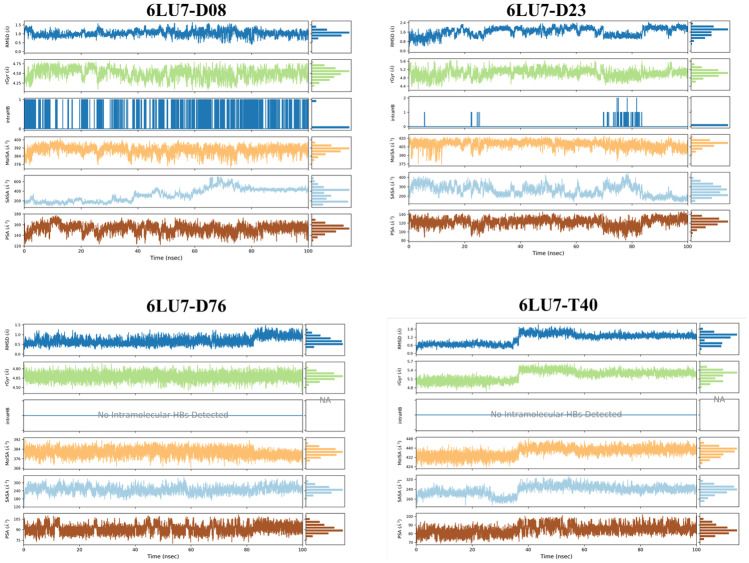
Small molecules such as 9,10-dihydrophenanthrene derivatives have remarkable activity toward inhibition of SARS-CoV-2 3CL and COVID-19 proliferation, which show a strong correlation between their structures and bioactivity. Therefore, these small compounds could be suitable for clinical pharmaceutical use against COVID-19. The objective of this study was to remodel the structures of 9,10-dihydrophenanthrene derivatives to achieve a powerful biological activity against 3CL and favorable pharmacokinetic properties for drug design and discovery. Therefore, by the use of bioinformatics techniques, we developed robust 3D-QSAR models that are capable of describing the structure-activity relationship for 46 molecules based on 9,10-dihydrophenanthrene derivatives using CoMFA/SE ( = 0.97, = 0.81, = 0.95, = 0.71) and CoMSIA/SEHDA ( = 0.94, = 0.76, = 0.91, = 0.65) techniques. Accordingly, 96 lead compounds were generated based on a template molecule that showed the highest observed activity in vitro (T40, pIC = 5.81) and predicted their activities and bioavailability in silico. The rational screening outputs of 3D-QSAR, Molecular docking, ADMET, and MM-GBSA led to the identification of 9 novel modeled molecules as potent noncovalent drugs against SARS-CoV-2-3CL. Finally, by molecular dynamics simulations, the stability and structural dynamics of 3CL free and complex (PDB code: 6LU7) were discussed in the presence of samples of 9,10-dihydrophenanthrene derivative in an aqueous environment. Overall, the retrosynthesis of the proposed drug compounds in this study and the evaluation of their bioactivity in vitro and in vivo may be interesting for designing and discovering a new drug effective against COVID-19.
The online version contains supplementary material available at 10.1007/s11224-022-02004-z.
9,10 - 二氢菲衍生物等小分子对抑制严重急性呼吸综合征冠状病毒2(SARS-CoV-2)3C样蛋白酶(3CL)和新型冠状病毒肺炎(COVID-19)增殖具有显著活性,其结构与生物活性之间存在很强的相关性。因此,这些小分子化合物可能适合用于抗COVID-19的临床药物应用。本研究的目的是对9,10 - 二氢菲衍生物的结构进行重塑,以获得针对3CL的强大生物活性以及有利于药物设计和发现的药代动力学性质。因此,通过使用生物信息学技术,我们基于比较分子场分析/表面静电(CoMFA/SE,R² = 0.97,q² = 0.81,Rpred² = 0.95,F = 0.71)和比较分子相似性指数分析/表面静电氢键供体和受体(CoMSIA/SEHDA,R² = 0.94,q² = 0.76,Rpred² = 0.91,F = 0.65)技术,开发了能够描述基于9,10 - 二氢菲衍生物的46个分子的构效关系的稳健三维定量构效关系(3D-QSAR)模型。据此,基于在体外表现出最高观察活性的模板分子(T40,pIC = 5.81)生成了96个先导化合物,并在计算机模拟中预测了它们的活性和生物利用度。3D-QSAR、分子对接、药物代谢及药物相互作用(ADMET)和分子力学广义Born表面面积(MM-GBSA)的合理筛选结果导致鉴定出9个新型建模分子作为针对SARS-CoV-2-3CL的强效非共价药物。最后,通过分子动力学模拟,在水环境中存在9,10 - 二氢菲衍生物样品的情况下,讨论了游离3CL和复合物(蛋白质数据银行代码:6LU7)的稳定性和结构动力学。总体而言,本研究中所提出的药物化合物的逆合成以及它们在体外和体内的生物活性评估对于设计和发现一种有效的抗COVID-19新药可能是有意义的。
在线版本包含可在10.1007/s11224-022-02004-z获取的补充材料。