Lewin Simon, Francioli Davide, Ulrich Andreas, Kolb Steffen
Microbial Biogeochemistry, Research Area Landscape Functioning, Leibniz Centre for Agricultural Landscape Research e.V. (ZALF), Müncheberg, Germany.
Thaer Institute, Faculty of Life Sciences, Humboldt University of Berlin, Berlin, Germany.
Environ Microbiome. 2021 Oct 12;16(1):18. doi: 10.1186/s40793-021-00387-w.
The native crop bacterial microbiota of the rhizosphere is envisioned to be engineered for sustainable agriculture. This requires the identification of keystone rhizosphere Bacteria and an understanding on how these govern crop-specific microbiome assembly from soils. We identified the metabolically active bacterial microbiota (SSU RNA) inhabiting two compartments of the rhizosphere of wheat (Triticum aestivum L.), barley (Hordeum vulgare L.), rye (Secale cereale), and oilseed rape (Brassica napus L.) at different growth stages.
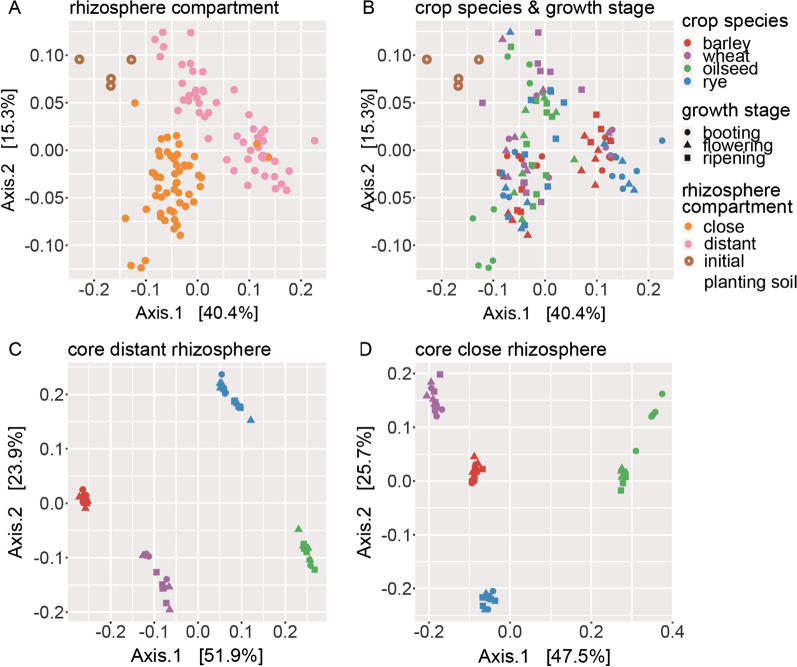
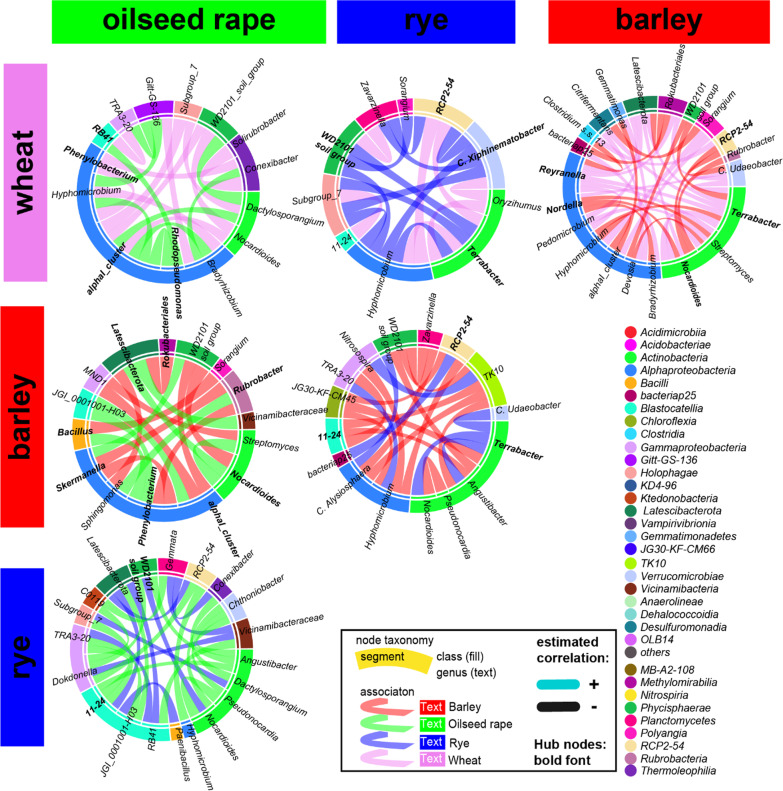
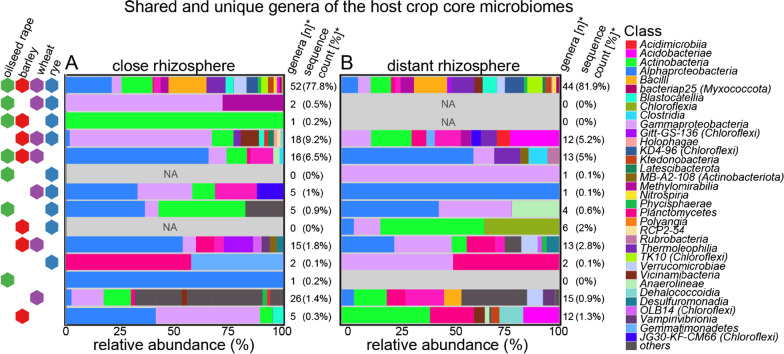
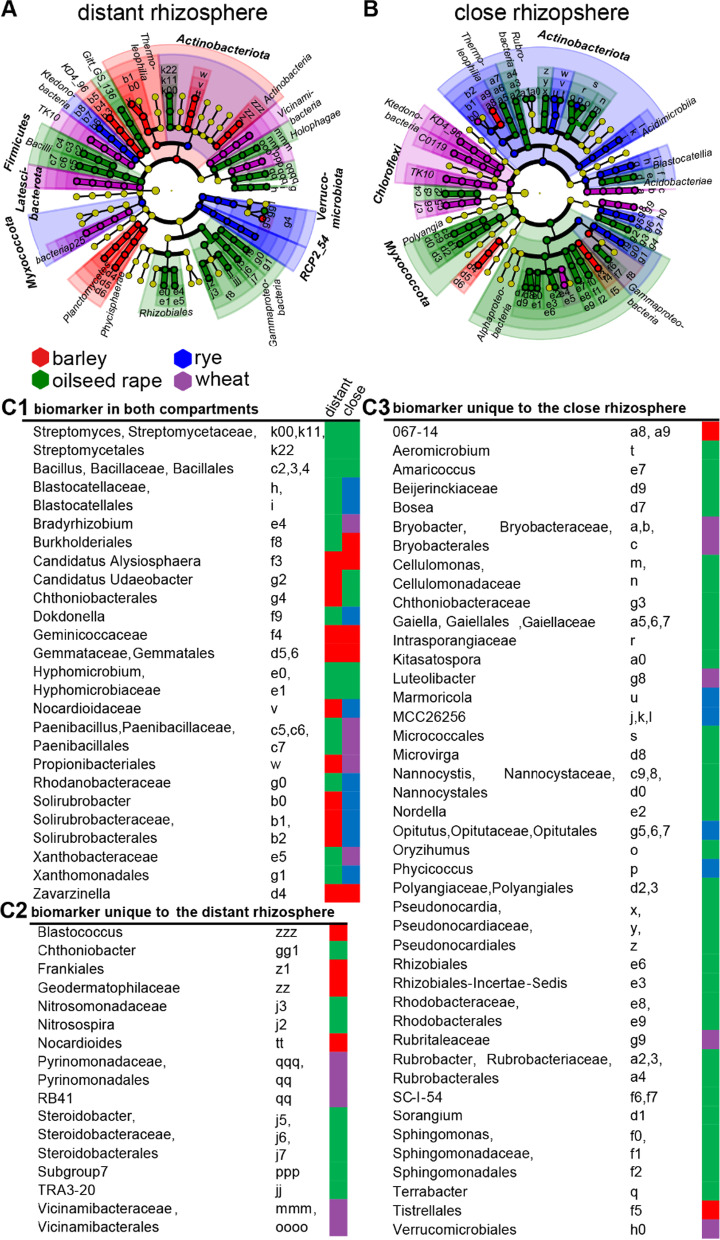
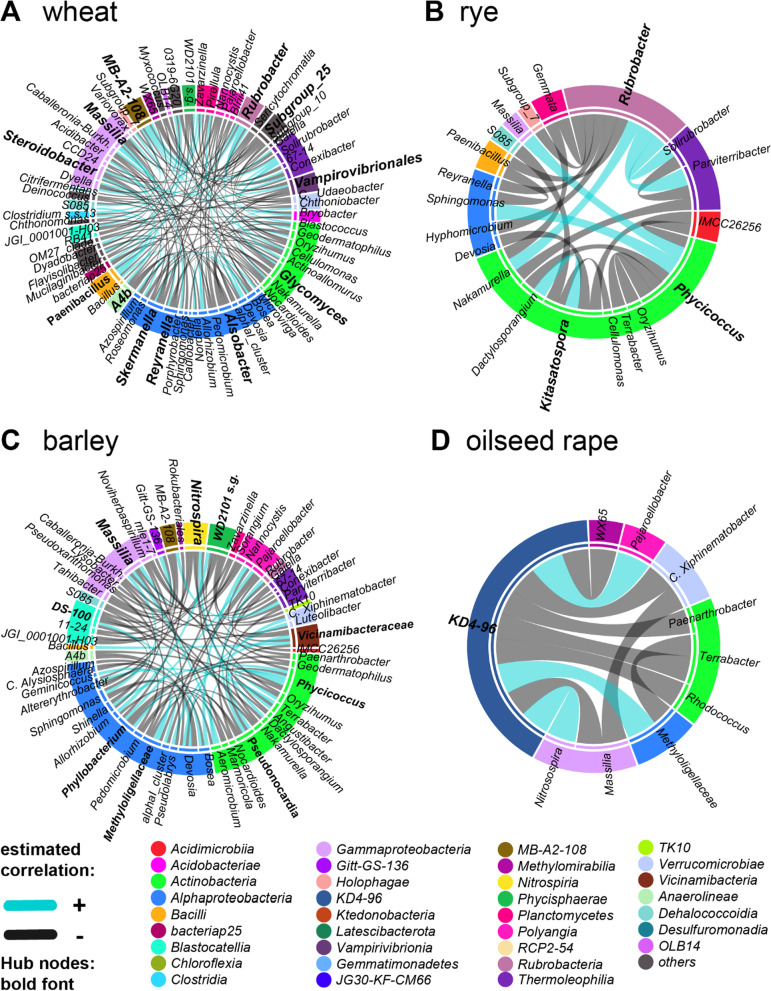
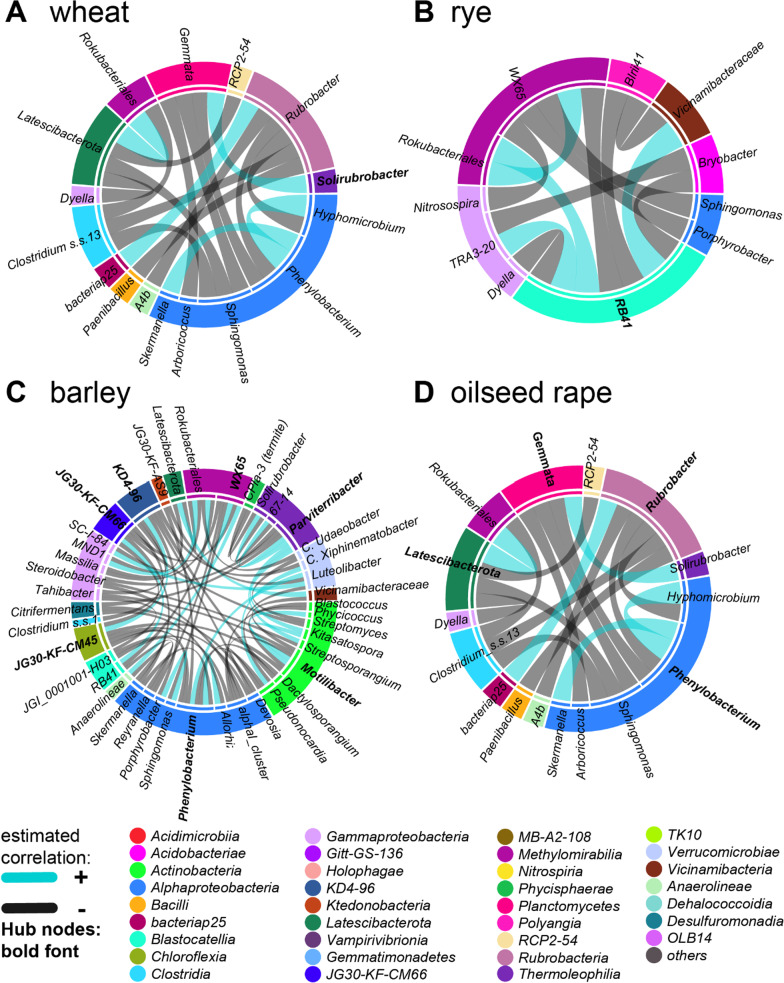
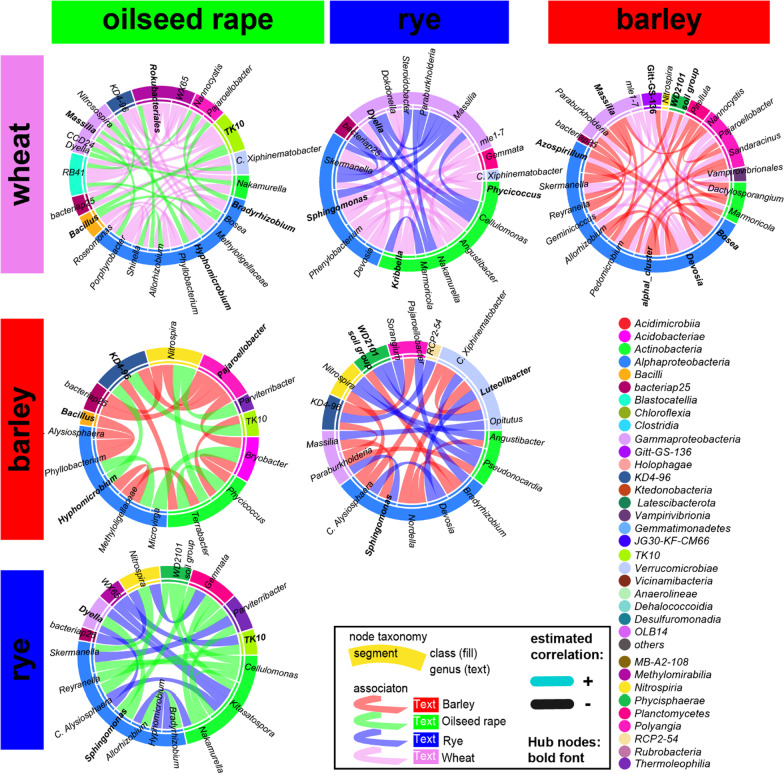
Based on metabarcoding analysis the bacterial microbiota was shaped by the two rhizosphere compartments, i.e. close and distant. Thereby implying a different spatial extent of bacterial microbiota acquirement by the cereals species versus oilseed rape. We derived core microbiota of each crop species. Massilia (barley and wheat) and unclassified Chloroflexi of group 'KD4-96' (oilseed rape) were identified as keystone Bacteria by combining LEfSe biomarker and network analyses. Subsequently, differential associations between networks of each crop species' core microbiota revealed host plant-specific interconnections for specific genera, such as the unclassified Tepidisphaeraceae 'WD2101 soil group'.
Our results provide keystone rhizosphere Bacteria derived from for crop hosts and revealed that cohort subnetworks and differential associations elucidated host species effect that was not evident from differential abundance of single bacterial genera enriched or unique to a specific plant host. Thus, we underline the importance of co-occurrence patterns within the rhizosphere microbiota that emerge in crop-specific microbiomes, which will be essential to modify native crop microbiomes for future agriculture and to develop effective bio-fertilizers.
人们设想对根际原生作物细菌微生物群进行改造,以实现可持续农业。这需要识别关键的根际细菌,并了解它们如何控制土壤中特定作物微生物群的组装。我们鉴定了在不同生长阶段栖息于小麦(Triticum aestivum L.)、大麦(Hordeum vulgare L.)、黑麦(Secale cereale)和油菜(Brassica napus L.)根际两个区域的代谢活跃细菌微生物群(SSU RNA)。
基于元条形码分析,细菌微生物群受根际两个区域(即近根区和远根区)的影响。这意味着谷类作物与油菜获取细菌微生物群的空间范围不同。我们得出了每种作物的核心微生物群。通过结合线性判别分析效应大小(LEfSe)生物标志物和网络分析,鉴定出马赛菌属(大麦和小麦)以及“KD4 - 96”组未分类的绿弯菌门(油菜)为关键细菌。随后,每种作物核心微生物群网络之间的差异关联揭示了特定属的宿主植物特异性相互联系,例如未分类的温泉微菌科“WD2101土壤群”。
我们的研究结果提供了源自作物宿主的关键根际细菌,并表明群组子网和差异关联阐明了宿主物种效应,而这从特定植物宿主富集或独特的单个细菌属的差异丰度中并不明显。因此,我们强调了根际微生物群中出现的共现模式在特定作物微生物群中的重要性,这对于未来农业改造原生作物微生物群和开发有效的生物肥料至关重要。