Department of Electrical and Computer Engineering, Carnegie Mellon University, Pittsburgh, PA, 15213, USA.
Department of Mechanical Engineering, Carnegie Mellon University, Pittsburgh, PA, 15213, USA.
Comput Biol Med. 2021 Nov;138:104915. doi: 10.1016/j.compbiomed.2021.104915. Epub 2021 Oct 5.
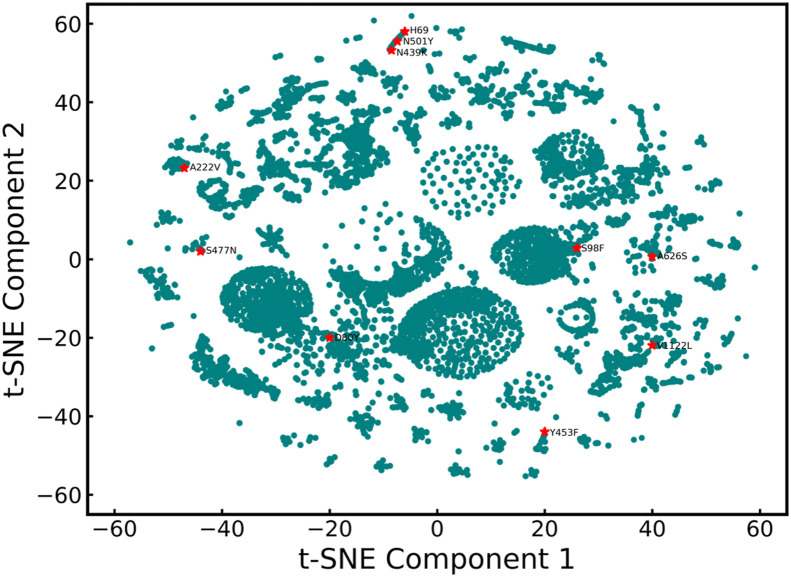
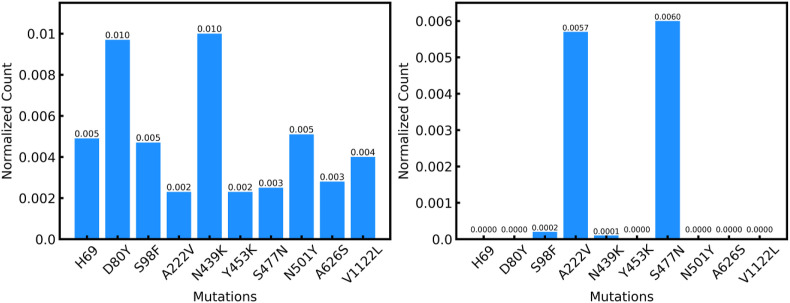
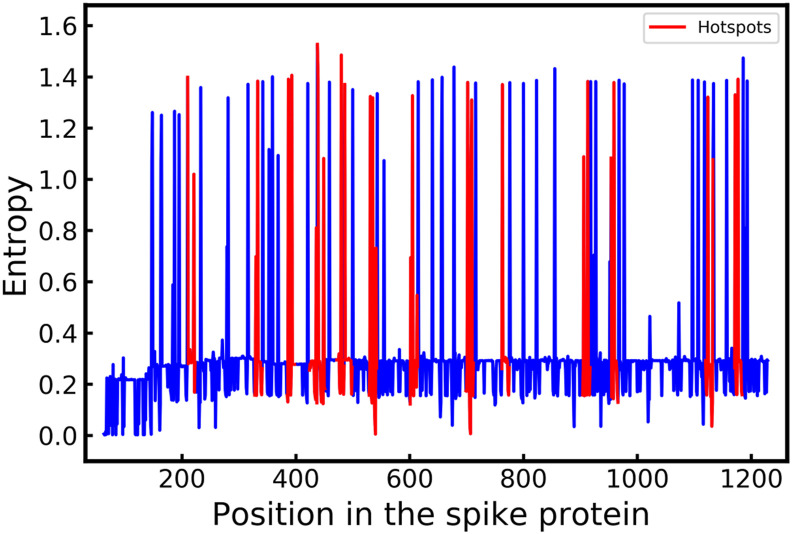
The SARS-CoV-2 virus like many other viruses has transformed in a continual manner to give rise to new variants by means of mutations commonly through substitutions and indels. These mutations in some cases can give the virus a survival advantage making the mutants dangerous. In general, laboratory investigation must be carried to determine whether the new variants have any characteristics that can make them more lethal and contagious. Therefore, complex and time-consuming analyses are required in order to delve deeper into the exact impact of a particular mutation. The time required for these analyses makes it difficult to understand the variants of concern and thereby limiting the preventive action that can be taken against them spreading rapidly. In this analysis, we have deployed a statistical technique Shannon Entropy, to identify positions in the spike protein of SARS Cov-2 viral sequence which are most susceptible to mutations. Subsequently, we also use machine learning based clustering techniques to cluster known dangerous mutations based on similarities in properties. This work utilizes embeddings generated using language modeling, the ProtBERT model, to identify mutations of a similar nature and to pick out regions of interest based on proneness to change. Our entropy-based analysis successfully predicted the fifteen hotspot regions, among which we were able to validate ten known variants of interest, in six hotspot regions. As the situation of SARS-COV-2 virus rapidly evolves we believe that the remaining nine mutational hotspots may contain variants that can emerge in the future. We believe that this may be promising in helping the research community to devise therapeutics based on probable new mutation zones in the viral sequence and resemblance in properties of various mutations.
SARS-CoV-2 病毒与许多其他病毒一样,通过常见的替换和插入/缺失突变,不断地发生变异,从而产生新的变体。在某些情况下,这些突变可以使病毒获得生存优势,使突变体变得危险。一般来说,必须进行实验室研究,以确定新变体是否具有任何使其更具致命性和传染性的特征。因此,需要进行复杂且耗时的分析,以便更深入地研究特定突变的确切影响。这些分析所需的时间使得难以理解关注的变体,从而限制了针对它们快速传播的预防措施。在这项分析中,我们部署了一种统计技术——香农熵,以确定 SARS-CoV-2 病毒序列刺突蛋白中最容易发生突变的位置。随后,我们还使用基于机器学习的聚类技术,根据性质的相似性对已知的危险突变进行聚类。这项工作利用语言建模生成的嵌入,即 ProtBERT 模型,来识别具有相似性质的突变,并根据易变性质选择感兴趣的区域。我们基于熵的分析成功预测了 15 个热点区域,其中我们能够在 6 个热点区域中验证 10 个已知的感兴趣变体。由于 SARS-CoV-2 病毒的情况迅速演变,我们认为其余 9 个突变热点可能包含未来可能出现的变体。我们相信,这有助于研究界根据病毒序列中可能出现的新突变区域和各种突变的性质相似性,设计基于治疗的方法。